N° 39417-S
EL PRESIDENTE DE LA
REPÚBLICA
Y EL MINISTRO DE
SALUD
En
uso de las facultades que le confieren los artículos 140 incisos 3), 18) y 146)
de la Constitución Política; 11, 25, 27, 28, párrafo 2, inciso b) de la Ley Nº 6227
del 2 de mayo de 1978 "Ley General de la Administración Pública"; 1, 2, 4, 113
y 114, de la Ley N ° 5395 del 30 de octubre de 1973, "Ley General de Salud"; 1
y 2 inciso b) de la Ley N° 5412 del 8 de noviembre de 1973, "Ley Orgánica del
Ministerio de Salud"
Considerando:
1º-Que
conforme a las disposiciones contenidas en el artículo 1° de la Ley General de
Salud, la salud de la población es un bien de interés público tutelado por el
Estado.
2º-Que
al amparo de las disposiciones legales contenidas en el artículo 2 inciso b) de
la Ley Orgánica del Ministerio de Salud, son atribuciones del Ministerio dictar
las normas técnicas en materia de salud particular o general; y ordenar las
medidas y disposiciones que técnicamente procedan en resguardo de la salud de
la población.
3º-Que
en atención a lo establecido en el artículo 10 inciso f) del Decreto Ejecutivo
Nº 35244-S "Reglamento del Sistema Nacional de Farmacovigilancia", el Centro
Nacional de Farmacovigilancia procedió a la elaboración de las Buenas Prácticas
de Farmacovigilancia.
4º-Que
las Buenas Prácticas de Farmacovigilancia contribuyen a asegurar la calidad e
integridad de los datos obtenidos mediante las notificaciones de reacciones
adversas, como parte del seguimiento del balance beneficio/riesgo de los
medicamentos cuando son utilizados en la población general, lo cual permite
vigilar su idoneidad para las indicaciones y condiciones de uso aprobados. Por
tanto,
DECRETAN:
Artículo
1°-Aprobar el siguiente reglamento:
Reglamento
de Buenas Prácticas de Farmacovigilancia
CAPÍTULO I
Disposiciones
Generales
1. OBJETIVO GENERAL.
Definir
las bases que contribuyan a establecer un sistema de garantía de calidad en las
actividades del Sistema Nacional de Farmacovigilancia, mediante el
establecimiento de las obligaciones y responsabilidades que deben cumplir los
diferentes agentes que lo conforman, con el fin de garantizar criterios
uniformes para realizar la evaluación de las notificaciones, la generación de
alertas y fomentar la comprensión y la enseñanza de la Farmacovigilancia.
2.
ÁMBITO DE APLICACIÓN.
Las disposiciones del
presente reglamento se aplican a cada uno de los agentes que conforman el
Sistema Nacional de Farmacovigilancia y a todos los medicamentos de uso humano
que se importan, fabrican, comercializan y utilizan en el país.
3.
REFERENCIAS.
Para la adecuada
interpretación y aplicación del presente reglamento se deben consultar los
siguientes documentos:
3.1 Reglamento
del Sistema Nacional de Farmacovigilancia. Decreto Ejecutivo N° 35244-S, del 13
de abril de 2009, publicado en La Gaceta N° 98 del 22/05/2009.
3.2 Reglamento
Técnico Centroamericano RTCA 11.03.59:11 Productos Farmacéuticos. Medicamentos
para uso Humano. Requisitos de Registro Sanitario. Anexo 1 de la Resolución Nº
333-2013 (COMIECO-LXVI), publicado en el Alcance digital Nº 20 de La Gaceta Nº
103 del 30 de mayo del 2014.
3.3 Reglamento para la autorización para la
importación y adquisición de medicamentos no registrados. Decreto Ejecutivo Nº
36358-S, del 4 de octubre del 2010, publicado en La Gaceta Nº 25 del 04 de
febrero del 2011.
4.
ABREVIATURAS.
4.1
BPFV: Buenas Prácticas de Farmacovigilancia.
4.2
CIOMS: Consejo de Organizaciones Internacionales de las Ciencias
Médicas (Council for International Organizations of Medical Sciences).
4.3
CNFV: Centro Nacional de Farmacovigilancia.
4.4
DRPIS: Dirección de Regulación de Productos de Interés Sanitario.
4.5
ESAVI: Evento Supuestamente Atribuible a la Vacunación o
Inmunización.
4.6
FV: Farmacovigilancia.
4.7
IBD: Fecha Internacional de Nacimiento (International Birth Date).
4.8
ICH: Conferencia Internacional de Armonización (International
Conference on Harmonization).
4.9
IPS: Informe Periódico de Seguridad.
4.10
OMS: Organización Mundial de la Salud.
4.11
OPS: Organización Panamericana de la Salud.
4.12
PNT: Procedimientos Normalizados de Trabajo.
4.13
RAM: Reacción Adversa Medicamentosa.
4.14
SNFV: Sistema Nacional de Farmacovigilancia.
4.15
WHO-art: Terminología de reacción adverse de la Organización Mundial
de la Salud (The WHO Adverse reaction terminology).
5.
DEFINICIONES.
Para
efectos de interpretación del presente reglamento, se utilizan además de las
definiciones establecidas en el Decreto Ejecutivo N° 35244-S Reglamento del
Sistema Nacional de Farmacovigilancia, las siguientes:
5.1
Alerta: Información comunicada sobre una posible relación causal
entre un evento adverso y un medicamento, cuando previamente se desconocía esta
relación o estaba documentada en forma incompleta.
5.2
Auditoría: Revisión de actividades específicas efectuadas con la
finalidad de verificar el cumplimiento de las Buenas Prácticas de
Farmacovigilancia.
5.3
Base de datos de farmacovigilancia: Sistema informático que
permite registrar notificaciones de sospechas de reacciones adversas, una vez
evaluadas y codificadas.
5.4
Buenas Prácticas de Farmacovigilancia: Conjunto de reglas
destinadas a garantizar la autenticidad y la calidad de los datos recolectados
en Farmacovigilancia, que permitan evaluar en cada momento los riesgos
atribuibles al medicamento; la confidencialidad de la información que se ha
notificado sobre las reacciones adversas y el uso de criterios uniformes en la
evaluación de las notificaciones y en la generación de señales de alerta.
5.5
Causalidad: Relación entre la aparición del evento adverso reportado y el
consumo de un medicamento específico.
5.6
Confidencialidad: Respeto del secreto de la identidad de la persona de quien se
ha notificado una sospecha de reacción adversa y que se extiende a toda la
información de carácter personal o
clínico. De forma similar, se resguarda la confidencialidad
de la información personal relativa a los profesionales notificadores.
5.7
Crisis: Una crisis se produce cuando se da a conocer información
nueva sobre la seguridad o eficacia de un producto que puede tener un efecto
importante en la salud pública y que, por tanto, requiere acciones inmediatas.
También puede sobrevenir cuando los medios de comunicación difunden información
en la que se expresa alguna preocupación acerca del consumo de determinado
producto.
5.8
Eficacia: Aptitud de un medicamento para producir los efectos
propuestos.
5.9
Estudio post-comercialización: Cualquier estudio clínico o epidemiológico
realizado durante la comercialización de un medicamento según las condiciones
autorizadas en el registro sanitario, o bien en condiciones normales de uso, en
el que el medicamento o los medicamentos de interés son el factor de exposición
fundamental investigado.
5.10
Falla terapéutica: Toda aquella situación en que no se logre el efecto
terapéutico esperado en el paciente, bajo dosificaciones adecuadas según la
prescripción utilizada con fines profilácticos, diagnósticos, terapéuticos o
para modificar una función fisiológica.
5.11
Farmacovigilancia: Actividad de salud pública destinada a la identificación,
cuantificación, evaluación y prevención de los riesgos asociados del uso de
medicamentos de uso humano una vez comercializados.
5.12
Farmacovigilancia intensiva: Método de la
farmacovigilancia que consiste en obtener información de sospechas de
reacciones adversas a los medicamentos de manera sistemática, de calidad y
completa, caracterizada por su elevada sensibilidad y fiabilidad; especialmente
cuando se necesita determinar la frecuencia de las reacciones adversas e
identificar agentes predisponentes y patrones de uso de medicamentos, entre
otros.
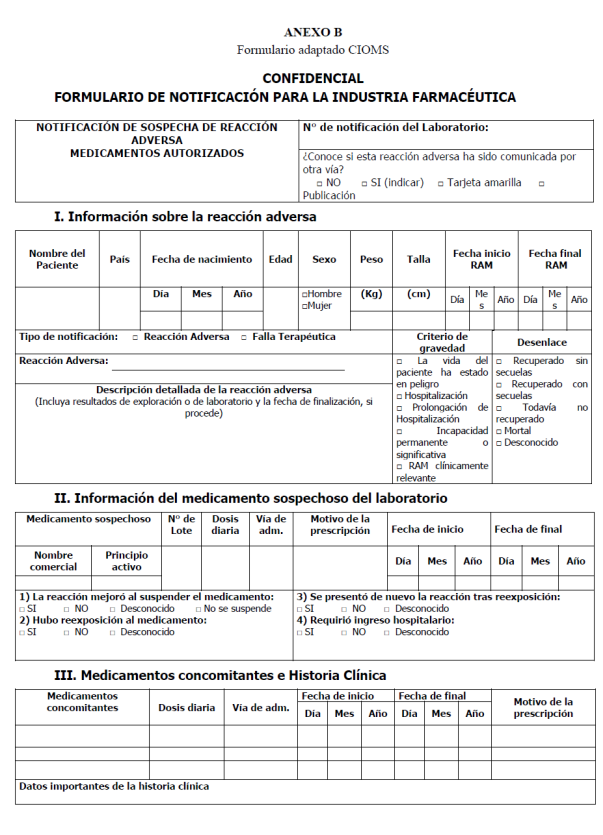
5.13
Formulario CIOMS adaptado: Es el formulario creado por CIOMS y
adaptado por el CNFV para el reporte de sospechas de reacciones adversas por
parte de la industria farmacéutica.
5.14
Hospital: Establecimiento de salud con al menos cinco camas para
internamiento de pacientes, que ofrece atención básica de diagnóstico y
tratamiento; cuerpo clínico organizado, con evidencia de admisiones y
asistencia permanente conducida de médicos.
5.15
Importación paralela: Importación de productos farmacéuticos patentados y
registrados en Costa Rica, por parte de cualquier droguería sin el
consentimiento del titular de la patente y que es comercializado conforme a las
regulaciones sanitarias del país exportador.
5.16
Indicación: Los usos a los cuales se destina un medicamento, después que
se ha probado científicamente que su empleo para una finalidad determinada es
efectivo y seguro.
5.17
Industria farmacéutica, titular del producto o titular del registro: Persona
física o jurídica propietaria del medicamento.
5.18
Informe periódico de seguridad: Documento preparado por el
titular del producto, cuya finalidad es actualizar la información de seguridad
del medicamento que, entre otros elementos, contiene información de las
sospechas de reacciones adversas de las que haya tenido conocimiento en el
período de referencia, así como una evaluación científica del balance
beneficio/riesgo del medicamento.
5.19
Monografía: Descripción científico técnica de un medicamento que debe
contener lo establecido en el numeral 7.6. del Reglamento Técnico
Centroamericano RTCA 11.03.59:11 Productos Farmacéuticos. Medicamentos Para Uso
Humano. Requisitos de Registro Sanitario:
a)
Denominación común o genérica internacionalmente aceptada y concentración del
medicamento.
b)
Forma farmacéutica.
c) Estructura, nombre químico del principio activo o en su
defecto adjuntar la ficha técnica que declare esta información.
d)
Farmacología clínica.
e)
Indicaciones.
f)
Contraindicaciones.
g)
Precauciones y advertencias.
h)
Interacciones.
i)
Efectos adversos.
j)
Dosis y administración.
k)
Recomendación en caso de sobredosificación según el perfil toxicológico.
l)
Abuso y adicción.
m)
Fecha de revisión de la monografía.
n)
Referencias bibliográficas completas.
o)
Clase terapéutica según Clasificación Anatómica Terapéutica (ATC).
p)
Forma de preparación.
5.20
Notificación: Comunicación de una sospecha de reacción adversa a un
medicamento al CNFV mediante los formularios de notificación de reacción
adversa (Tarjeta Amarilla o Formulario CIOMS modificado) establecidos por el
Ministerio de Salud.
5.21
Plan de minimización de riesgos: Documento en el que el
titular del producto especifica los riesgos asociados al medicamento,
identificados o potenciales y señala la información de seguridad no conocida en
la literatura científica. Consiste en un programa estratégico de seguridad
orientado a alcanzar metas y objetivos específicos para reducir al mínimo los
riesgos conocidos de los medicamentos preservando sus beneficios.
5.22
Procedimientos Normalizados de Trabajo: Instrucciones escritas y
detalladas para lograr la uniformidad en la realización de una actividad
específica. Son la base fundamental para las auditorías internas o externas.
5.23
Rastreabilidad (Trazabilidad): Posibilidad de encontrar y
seguir el rastro de un medicamento, a través de todas las etapas de producción,
almacenamiento, distribución y comercialización.
5.24
Riesgo: Probabilidad de ocasionar un perjuicio o daño a la salud como
resultado del uso de un medicamento, asociado a la magnitud del mismo.
5.25
Relación Beneficio/riesgo: Refleja la correlación entre el
beneficio y el riesgo que presenta el uso de un medicamento. Sirve para
expresar un juicio sobre la función del medicamento en la práctica médica,
basado en datos sobre su eficacia y seguridad y en consideraciones sobre su
posible uso indebido, la gravedad y el pronóstico de la enfermedad, etcétera.
El concepto puede aplicarse a un solo medicamento o a las comparaciones entre
dos o más medicamentos empleados para una misma indicación.
5.26
Señal: Posible relación causal entre un evento adverso y un
medicamento cuando previamente se desconocía esta relación o estaba documentada
en forma incompleta. Habitualmente se requiere más de una notificación para
generar una señal, dependiendo de la gravedad del evento adverso y de la calidad
de la información.
5.27
Sistema de notificación espontánea: Método de farmacovigilancia
basado en la comunicación, reporte, recolección y evaluación de notificaciones
de sospechas de reacciones
adversas a medicamentos realizadas por un profesional de la
salud a través de los formularios establecidos.
5.28
WHo-art: Diccionario de reacciones adversas de la Organización Mundial
de la Salud, que contiene la terminología para codificar la información clínica
relacionada con los medicamentos.
6.
SISTEMA NACIONAL DE FARMACOVIGILANCIA.
El
SNFV está regulado por medio del Decreto Ejecutivo Nº 35244-S "Reglamento del
Sistema Nacional de Farmacovigilancia", el cual aplica a la FV de todos los
medicamentos de uso humano que se importan, fabrican, comercializan y utilizan
en el país.
7.
FUNCIONAMIENTO DEL SISTEMA NACIONAL DE FARMACOVIGILANCIA.
7.1 Los
profesionales en salud deben enviar las notificaciones al CNFV en los
formularios designados para la recolección de los datos.
7.2 La
información de seguridad debe ser evaluada periódicamente por el CNFV según el
PNT establecido con el fin de identificar de forma temprana posibles problemas
de seguridad derivados del uso de los medicamentos para la generación de
señales.
7.3 Las
señales serán discutidas y analizadas en la Comisión Nacional de
Farmacovigilancia, atendiendo al PNT establecido.
7.4 La
Comisión Nacional de Farmacovigilancia debe asesorar a la DRPIS para la toma de
decisiones.
7.5 La
DRPIS debe tomar las medidas necesarias para mantener la relación beneficio/riesgo
favorable de los medicamentos.
7.6 La
DRPIS debe informar a los profesionales y al público en general sobre las
medidas de seguridad adoptadas para mantener dicha relación beneficio/riesgo
favorable.
CAPÍTULO II
Sobre
la Información del Sistema Nacional de Farmacovigilancia
8.
DOCUMENTACIÓN.
Los
Agentes del SNFV deben contar con documentación completa y actualizada de
acuerdo a lo establecido en el presente reglamento.
8.1
Formularios de reporte.
8.1.1
Todos los profesionales en salud deben realizar la
notificación de sospechas de reacciones adversas utilizando el formulario
oficial Tarjeta Amarilla, adjunto en el Anexo A del presente reglamento y
procurando que la información suministrada esté completa.
8.1.2
La industria farmacéutica debe realizar la notificación de
sospechas de reacciones adversas a través del formulario adaptado CIOMS,
adjunto en el Anexo B del presente reglamento y procurando que la información
suministrada esté completa.
8.1.3
Tanto la industria farmacéutica como los profesionales en
salud deben notificar las sospechas de falla terapéutica a través de los
formularios mencionados en los incisos 8.1.1 y 8.1.2 incluidos en los Anexos A
y B del presente reglamento.
8.1.4
Para realizar el análisis de una notificación se requieren al
menos los siguientes datos reportados con letra legible y tinta indeleble:
a)
Paciente identificable: nombre o iniciales del paciente o cédula de identidad o
género.
b)
Medicamento sospechoso: nombre genérico o marca comercial.
c)
Fecha exacta o aproximada de inicio de la administración del medicamento.
d) Reacción adversa incluyendo la localización anatómica y
severidad (leve, moderada, grave o mortal), si se cuenta con ellas.
e)
Fecha exacta o aproximada de inicio de la reacción.
f)
Notificador identificable: nombre, firma y código profesional, teléfono de
contacto y dirección de correo electrónico.
8.1.5
En caso de contar con información complementaria, los
notificadores la deben documentar, enviar y conservar con el fin de que permita
ampliar la información contenida en el formulario de notificación de sospechas
de reacciones adversas. En el caso de que la información sea confidencial, se
debe incluir en el formulario un resumen de la información obtenida.
8.1.6
La notificación debe ser entregada en forma física al CNFV a
través de la Dirección de Atención al Cliente. La información puede ser enviada
por fax, correo electrónico o puede ser comunicada por vía telefónica al CNFV
con posterior entrega de la documentación física de acuerdo al Reglamento del
Sistema Nacional de Farmacovigilancia.
8.2
Procedimientos Normalizados de Trabajo.
8.2.1
El CNFV debe contar con PNT establecidos para cada una de sus actividades.
8.2.2
Los centros prestadores de servicios de salud y la industria farmacéutica deben
contar con PNT para cada actividad de FV que realicen, los cuales deben ser
revisados y aprobados por los encargados de FV, estar implementados y ser del
conocimiento por parte del personal involucrado.
CAPÍTULO III
Obligaciones
y Responsabilidades de los Agentes del SNFV
9.
CENTRO NACIONAL DE FARMACOVIGILANCIA.
El
CNFV debe cumplir con las siguientes obligaciones y responsabilidades:
9.1 Recibir,
evaluar, analizar y codificar las notificaciones de sospechas de RAM.
9.2 Vigilar
la seguridad de los medicamentos a través del análisis de las señales.
9.3 Comunicar
a la DRPIS toda información de seguridad de los medicamentos de uso humano
detectada durante los análisis realizados.
9.4 Divulgar
información sobre FV a los pacientes y a los profesionales de la salud.
9.5 Coordinar
las actividades de FV que realicen los diferentes agentes del SNFV para lograr
captar información necesaria y oportuna sobre las sospechas de RAM.
9.6 Participar
en actividades de capacitación en el tema de Farmacovigilancia dirigidas
principalmente a profesionales en salud y estudiantes universitarios en el área
de la salud.
9.7 Realizar
inspecciones a la labor del profesional encargado de FV y a la industria farmacéutica
con el fin de comprobar el cumplimiento de las BPFV y evaluar su eficacia para
alcanzar los objetivos específicos. Para facilitar las inspecciones de FV, el
Ministerio de Salud mantendrá a disposición de los administrados la Guía de
Verificación de BPFV disponible en la página web del Ministerio.
9.8 Identificar
las señales generadas, analizarlas y realizar investigaciones que permitan
concluir o descartar que el medicamento sea el causante del evento. Estas
señales pueden ser detectadas principalmente por los siguientes métodos de FV:
a)
Descripciones aisladas de pacientes.
b)
Publicación de casos en la literatura científica.
c)
Notificación espontánea al Sistema de Farmacovigilancia.
d)
Estudios observacionales en poblaciones: estudios de cohorte o de casos y
controles.
e) Estudios experimentales: investigaciones biomédicas.
Es
posible que un solo caso notificado, bien documentado, pueda verse como una
señal, sobre todo si describe una reexposición positiva o si el evento es
desconocido en ausencia del medicamento usado.
9.9 Evaluar
periódicamente la información contenida en la base de datos de FV del CNFV con
el fin de detectar señales, las cuales serán evaluadas y analizadas. Cuando se
considere que la señal detectada constituye un problema inminente de salud
pública, se debe realizar una investigación y un informe para la toma de
medidas sanitarias.
9.10
Cuantificar la fuerza de la asociación entre la reacción
adversa y el medicamento, una vez identificado un riesgo y su efecto en términos
de salud pública.
9.11
Evaluar los posibles beneficios y riesgos de los medicamentos
para los cuales se ha cuantificado un riesgo y velar porque la relación
beneficio/riesgo del medicamento siga siendo favorable.
9.12
Realizar estrategias para prevenir y minimizar los riesgos
asociados a los medicamentos, dentro de las cuales se encuentran:
a)
Ejecutar programas de FV Intensiva o de seguimiento sobre determinados
medicamentos o grupos de riesgo.
b)
Establecer mecanismos de integración de las actividades de vigilancia sanitaria
en materia de promoción y publicidad, en relación con la información sobre las
reacciones adversas, las advertencias y precauciones y las contraindicaciones.
c)
Realizar en forma sistemática y periódica la prevención de riesgos. Los
profesionales de la salud, los usuarios, la industria farmacéutica, los centros
prestadores de servicios de salud, el CNFV y la DRPIS tienen responsabilidades
compartidas.
Con
respecto a las reacciones adversas no evitables, el objetivo debe ser su detección
precoz como principal medida de prevención que reducirá la magnitud del daño.
9.13
Trabajar en forma articulada con los programas de salud
pública incluyendo el de inmunizaciones, de forma tal que las notificaciones de
eventos y sospechas de RAM detectadas a través de esos programas sean
notificadas al CNFV para su evaluación. Los ESAVIS, aunque se remitan a otras
instancias de salud pública, deben ser comunicados al CNFV en la Tarjeta
Amarilla según los lineamientos establecidos para el manejo de ESAVIS.
10.
CENTROS PRESTADORES DE SERVICIOS DE SALUD.
Todos
los centros prestadores de servicios de salud, es decir aquellos hospitales
públicos y privados así como las áreas de salud de la CCSS deben contar con un
encargado de FV.
10.1
El director médico del centro prestador de servicios de salud
debe designar y comunicar por escrito al CNFV el profesional de la salud
encargado de FV y sus datos de contacto. Asimismo debe gestionar los recursos
necesarios para que este encargado lleve a cabo de manera adecuada sus
responsabilidades.
10.2
El encargado de FV debe cumplir con los siguientes
requisitos, obligaciones y responsabilidades:
a)
Servir de contacto con el CNFV.
b) Tener
acceso a un teléfono, fax, computadora con internet, correo electrónico y
fotocopiadora.
c)
Impulsar el Sistema de Notificación Espontánea de RAM y otros programas de
conformidad con las BPFV en el centro prestador de servicios de salud al cual
pertenece.
d)
Distribuir el formulario de notificación de sospechas de RAM (Tarjeta Amarilla)
a los Profesionales en Salud dentro de su centro prestador de servicios de
salud.
e) Recibir las notificaciones de sospechas de RAM generadas
dentro de su centro prestador de servicios de salud con el único fin de ser
remitidas al CNFV.
f)
Llevar un registro de las notificaciones que recibe.
g)
Verificar que los datos de las Tarjetas Amarillas estén completos, en caso
contrario realizar las gestiones necesarias para completar los datos.
h)
Resguardar la confidencialidad de las notificaciones realizadas por los
profesionales en salud.
i)
Transferir las consultas o solicitudes de información relacionadas con
sospechas de RAM reacciones adversas formuladas por profesionales de salud de
su centro al CNFV.
j)
Responder a las solicitudes de información que le realice el CNFV.
k)
Coordinar en conjunto con el CNFV actividades de capacitación a profesionales
en salud.
l)
Participar en las reuniones, capacitaciones y otras actividades que el CNFV
programe.
m)
Establecer los PNT necesarios para garantizar las BPFV en su centro prestador
de servicios de salud.
11.
PROFESIONALES EN CIENCIAS DE LA SALUD.
11.1.
Los profesionales en ciencias de la salud deben cumplir con
los siguientes requisitos, obligaciones y responsabilidades:
a)
Participar activamente del SNFV para lograr captar de forma efectiva y oportuna
toda la información referente a las sospechas de RAM, las cuales tienen
implicación directa en la seguridad de los medicamentos que se utilizan en el
país.
b)
Cumplir las obligaciones establecidas en el Decreto Ejecutivo Nº 35244-S
"Reglamento del Sistema Nacional de Farmacovigilancia" en su artículo 11.
c)
Llenar la Tarjeta Amarilla de forma completa cuando detecte una sospecha de
RAM.
d)
Enviar las sospechas de RAM al CNFV en formularios oficiales y los plazos
establecidos en el Decreto Ejecutivo Nº 35244-S. Este envío se puede realizar a
través de las Direcciones de Áreas Rectoras de Salud del Ministerio de Salud.
e)
Resguardar la confidencialidad de las notificaciones realizadas al CNFV.
f)
Responder a las solicitudes de información que le realice el CNFV.
g)
Participar en las reuniones, capacitaciones y otras actividades que el CNFV
programe.
h)
Mantenerse informado sobre los datos de seguridad relativos a los medicamentos
que habitualmente prescriban, dispensen o administren.
i)
Colaborar, en caso necesario, en calidad de expertos con el CNFV en la
evaluación de los problemas de seguridad de los medicamentos.
11.2.
Los regentes farmacéuticos de las droguerías que realizan importación
de medicamentos mediante la modalidad de importación paralela y mediante la
aplicación del artículo 117 de la Ley General de Salud, en particular el
artículo 3 del Decreto Ejecutivo Nº 36358-S deben cumplir, además de los
requisitos mencionados en el numeral anterior, los siguientes requisitos,
obligaciones y responsabilidades:
a)
Velar porque se establezcan y se cumplan los PNT para asegurar la FV de los
productos importados mediante la modalidad indicada en el presente numeral.
b)
Garantizar que todo el personal que trabaja en la droguería tenga conocimiento
de los PNT.
c)
Ser el encargado del Programa de Farmacovigilancia o delegar tal función en un
profesional de la salud capacitado, el cual será la persona de contacto con el
CNFV.
d) Establecer acuerdos en materia de FV en caso de existir
cualquier transferencia de obligaciones y funciones. Estos deben estar
documentados mediante un acuerdo escrito firmado y legalizado entre los
representantes legales de las dos empresas, los cuales deben ser notificados al
CNFV. Las funciones no transferidas mediante este acuerdo siguen siendo
asumidas por el responsable de la importación.
e)
Gestionar toda medida sanitaria que le sea solicitada por la DRPIS en materia
de seguridad de los medicamentos.
f)
Llevar un registro detallado de las sospechas de RAM detectadas que incluya
toda la información contenida en el Formulario de Notificación de Sospechas de
RAM. Tal registro debe mantenerse en un sistema de archivo ya sea físico o
digital que permita conservar adecuadamente toda la documentación relacionada
con las responsabilidades y actividades de FV por un periodo de 5 años
g)
Responder en un plazo máximo de 10 días hábiles a cualquier solicitud de
información de la DRPIS en materia de seguridad de medicamentos.
h)
Evaluar de forma continua la relación beneficio/riesgo de los medicamentos y
comunicar en un plazo máximo de 10 días hábiles a la DRPIS sobre nueva
información de seguridad.
i)
Identificar señales y valorar la gravedad de las mismas, las cuales deben ser
comunicadas al CNFV.
j)
Realizar FV intensiva a los medicamentos cuando el CNFV así lo requiera. Para
facilitar el cumplimiento de los requisitos de FV intensiva, el Ministerio de
Salud mantendrá a disposición de los administrados la Guía para realizar FV
Intensiva en su página web.
12.
INDUSTRIA FARMACÉUTICA.
Todos
los titulares de registro de los medicamentos deben contar con un Programa de
Farmacovigilancia. Dicho programa debe contener los roles y responsabilidades
relacionadas con la seguridad de los medicamentos que comercializa y asegurar
la adopción de las medidas oportunas cuando sea necesario.
12.1
Obligaciones y responsabilidades del titular del registro:
a)
Garantizar que todo el personal que trabaja en la empresa tenga conocimiento en
materia de Farmacovigilancia.
b)
Contar con un profesional de la salud encargado del Programa de
Farmacovigilancia, el cual será la persona de contacto con el CNFV.
c)
Facilitar al profesional encargado del Programa de Farmacovigilancia el acceso
a la monografía e información básica de seguridad actualizadas de cada
medicamento.
d)
Establecer acuerdos en materia de FV. En caso de existir cualquier
transferencia de obligaciones y funciones, debe estar documentada mediante un
acuerdo escrito firmado y legalizado entre los representantes legales de las
dos empresas. Estos acuerdos deben ser notificados al CNFV y además deben
adjuntarse al expediente de registro sanitario. Las funciones no transferidas
mediante este acuerdo siguen siendo asumidas por el titular del registro.
e)
Velar porque se establezcan y se cumplan los PNT para asegurar la FV.
f)
Garantizar un sistema de archivo ya sea físico o digital que permita conservar
adecuadamente toda la documentación relacionada con las responsabilidades y
actividades de FV por un periodo de 5 años. Las responsabilidades en la gestión
del archivo deben estar definidas por escrito.
g)
Establecer un programa de auditorías internas, con el fin de garantizar que el
Programa de Farmacovigilancia cumpla con lo establecido en el presente
reglamento.
h)
Enviar al CNFV cualquier información relacionada con la seguridad de sus
medicamentos.
i) Remitir al CNFV (previo a su distribución) todo comunicado
relacionado con la seguridad de sus medicamentos que se desee divulgar a los
profesionales en salud o al público en general. El CNFV podrá solicitar
ampliaciones o modificaciones al comunicado.
j)
Todo comunicado relacionado con la seguridad de sus medicamentos debe indicar
la siguiente leyenda: "Toda sospecha de reacción adversa se debe notificar al
CNFV en los formularios y plazos establecidos en la normativa vigente".
12.2
Obligaciones y responsabilidades del encargado de FV:
a)
Notificar las sospechas de RAM, retiro del mercado por motivos de seguridad u otro
hecho relacionado con la seguridad de los medicamentos comercializados a nivel
nacional o internacional al CNFV.
b)
Gestionar toda medida sanitaria que le sea solicitada por la DRPIS en materia
de seguridad de los medicamentos.
c)
Llevar un registro detallado que incluya toda la información contenida en el
Formulario de Notificación de Sospechas de RAM.
d)
Remitir los IPS al CNFV. Para facilitar el cumplimiento de los requisitos para
los IPS, en cuanto a contenido, el Ministerio de Salud mantendrá a disposición
de los administrados la Guía de presentación de IPS para la Industria
Farmacéutica en su página web.
e)
Responder en un plazo máximo de 10 días hábiles a cualquier solicitud de
información de la DRPIS en materia de seguridad de medicamentos.
f)
Evaluar de forma continua la relación beneficio/riesgo de los medicamentos
comercializados en el país y comunicar en un plazo máximo de 10 días hábiles a
la DRPIS sobre nueva información de seguridad.
g)
Identificar señales y valorar la gravedad de las mismas, las cuales deben ser
comunicadas al CNFV.
h)
Realizar FV intensiva a los medicamentos cuando el CNFV así lo requiera. Para
facilitar el cumplimiento de los requisitos de FV intensiva, el Ministerio de
Salud mantendrá a disposición de los administrados la Guía para realizar FV
Intensiva en su página web.
i)
Participar en las reuniones, capacitaciones y otras actividades que el CNFV
programe.
j)
Colaborar, en caso necesario, en calidad de expertos con el CNFV en la
evaluación de los problemas de seguridad de los medicamentos.
k)
En caso de tener conocimiento de una sospecha de RAM relacionada con
medicamentos que no sean propios de su industria, debe informar al titular de
ese producto.
12.3
Organización y personal:
La
Industria Farmacéutica debe disponer de un organigrama actualizado en que se
refleje la relación jerárquica que hay entre el Encargado de FV, la Dirección
Médica y el resto de los departamentos y debe cumplir con los siguientes
aspectos:
a)
Debe existir una persona designada como encargada de FV y un suplente, ambos
con formación y experiencia en FV.
b)
El personal de FV debe conocer las funciones y responsabilidades asignadas, las
cuales tienen que estar por escrito, en las descripciones de los puestos de
trabajo, aprobadas por la dirección.
c)
El titular del registro debe mantener actualizado el curriculum vitae, la
descripción del puesto de trabajo y la capacitación del personal involucrado en
las tareas de FV.
d)
El titular del registro debe poner a disposición del encargado de FV los
recursos humanos y materiales necesarios para llevar a cabo de manera adecuada
sus responsabilidades.
12.4 Capacitación en FV al personal del titular del registro:
12.4.1
Se debe disponer de un programa de formación inicial y
continua en materia de FV, el cual debe ser aprobado por la persona encargada
de FV.
12.4.2
Todo el personal de la compañía relacionado con la recepción
de sospechas de RAM debe recibir formación inicial y continua en materia de FV.
12.4.3
Se deben conservar los registros que validen la capacitación
del personal.
12.5
Procedimientos Normalizados de Trabajo (PNT):
El
titular del registro debe disponer de PNT aprobados por el encargado de FV y
por la Dirección Médica, que describan las funciones y actividades que se lleven
a cabo en materia de FV y deben cumplir con lo siguiente:
a)
Estar actualizados de acuerdo a la información científica y la legislación
vigente. Debe mantenerse un archivo histórico con las actualizaciones de los
PNT.
b)
El encargado, así como todas aquellas personas implicadas en el Programa de
Farmacovigilancia deben de llevar a cabo las funciones y tareas de acuerdo con
lo establecido en los PNT.
c)
Los PNT deben de estar a disposición del personal encargado de llevar a cabo las
funciones y tareas descritas en su puesto de trabajo; así como las guías y
normativas vigentes sobre FV.
12.6
Gestión de las notificaciones de sospechas de RAM:
12.6.1
La información registrada en las notificaciones de sospechas
de RAM tendrán carácter de declaración jurada; la veracidad de la información
podrá ser verificada por el Ministerio de Salud.
12.6.2
En el momento en que un colaborador del titular del registro
recibe información inicial o de seguimiento de una RAM, debe comunicarlo a más
tardar un día hábil posterior a la recepción de la información, al encargado de
FV y debe quedar constancia de la fecha de conocimiento de esta RAM por parte
del titular de registro.
12.6.3
El encargado de FV debe asegurarse que se registra, se fecha
y asigna un número de identificación correlativo único e inequívoco a cada
notificación de RAM recibida.
12.6.4
Para cualquier sospecha de RAM el encargado de FV debe
asegurarse que se recopile toda la información necesaria y debe evaluar los
siguientes criterios: gravedad, si está referenciada o no de acuerdo con la
información básica de seguridad del producto, si es esperada o inesperada de
acuerdo con la monografía, cumpliendo con los plazos de reporte de acuerdo a lo
establecido en el Decreto Ejecutivo N° 35244-S "Reglamento del Sistema Nacional
de Farmacovigilancia". Esta información debe anotarse en el formulario adaptado
CIOMS en la sección de descripción de la RAM.
12.6.5
El encargado de Farmacovigilancia debe asegurar que se
realice un seguimiento de la evolución y el desenlace de cada caso individual,
luego de realizar al menos 3 intentos de contacto al notificador, los cuales
deben quedar documentados. La información de seguimiento adicional que se
reciba quedará registrada y fechada de igual forma que la información inicial y
debe ser enviada al CNFV en el formulario adaptado CIOMS en la sección de
descripción de la RAM.
12.6.6
Todos los documentos y registros relacionados con una misma
RAM deben conservarse conjuntamente o bien de manera que pueda localizarse
fácilmente y se pueda hacer un seguimiento de todas las actividades
relacionadas con la detección, evaluación y notificación.
12.6.7
Cuando se reciba información directamente de un paciente, que
sugiera que se ha producido una RAM, el titular del registro debe intentar
obtener el permiso del paciente para contactar con el profesional sanitario
responsable del seguimiento clínico, con el fin de obtener información.
12.6.8
La información referente a sobredosis, exposición en embarazo
o lactancia, uso incorrecto, dependencia, abuso de medicamentos o a errores de
medicación deberá ser recolectada y enviada al CNFV, en el formulario adaptado
CIOMS en los plazos establecidos en el Decreto Ejecutivo N° 35244-S "Reglamento
del Sistema Nacional de Farmacovigilancia".
12.7 Gestión de los datos:
La
gestión de los datos se debe realizar de acuerdo a los siguientes puntos:
a)
El sistema de gestión de datos de sospechas de RAM debe asegurar la integridad,
exactitud, fiabilidad, consistencia y confidencialidad de toda la información.
b)
Cuando se realice cualquier corrección de datos debe hacerse de manera que
puedan leerse los datos anteriores, documentando el motivo del cambio, la fecha
e identificación (por ejemplo: firma, iniciales, código de usuario, etc) de la
persona que hizo la modificación.
c)
Si se transforman los datos durante su procesamiento, debe mantenerse su
rastreabilidad, de manera que se puedan comparar los datos iniciales con los
cambios sucesivos.
d)
El sistema de gestión de datos debe permitir la búsqueda rápida y selectiva de
información según criterios de gravedad, edad del paciente, sexo, fármacos,
fechas de notificación, fechas de la RAM, procedencia, etc. El sistema debe
permitir el acceso inmediato a los datos esenciales (número de identificación
del caso, fármaco, reacción y narrativa del caso).
e)
Si se transfiere información de FV de cualquier fuente interna (por ejemplo,
entre departamento médico y departamento de FV), o externa (entre la central y
filiales o entre compañías licenciatarias), deben establecerse procedimientos
de intercambio de información para garantizar la completa transferencia.
f)
Los sistemas informáticos de gestión de datos deben cumplir los siguientes
requisitos:
Disponer
de medidas físicas de seguridad que impidan el acceso no autorizado a los
equipos y soportes informáticos.
Implementar
medidas de seguridad que impidan el acceso no autorizado a los datos. Debe
existir una lista actualizada de las personas autorizadas con su
correspondiente nivel de acceso al sistema (de consulta, introducción,
modificación de datos, etc).
Realizar
regularmente copias de seguridad de los datos.
Documentar
y validar cualquier proceso de migración de datos a otro sistema.
Identificar
los datos registrados en el sistema informático con su autor, fecha y hora de
introducción. Debe existir un registro de datos que permita dar seguimiento y
conocer todos los cambios secuenciales asociados a un dato, con identificación
de su autor, fecha, hora y valor anterior. Cualquier cambio debe estar
justificado.
Disponer
de un procedimiento alternativo de gestión de datos en caso de fallo temporal
del sistema, que permita garantizar el cumplimiento de las obligaciones legales
de FV.
Establecer
un plan de recuperación ante desastres en caso de fallo permanente del sistema.
g)
El titular del registro debe garantizar una adecuada formación y entrenamiento
del personal, adaptados a sus responsabilidades en el uso de los sistemas
informáticos de gestión de datos.
h) Debe
disponer de PNT que describan todas las actividades relacionadas con los
sistemas informáticos.
12.8.
Informes Periódicos de Seguridad (IPS):
12.8.1
La presentación de los IPS se debe realizar de acuerdo a los
siguientes puntos:
a)
La presentación es obligatoria para todos los medicamentos innovadores, los
medicamentos biológicos incluyendo a los biosimilares, así como para los
medicamentos que deben demostrar la equivalencia terapéutica.
b)
El titular del registro debe elaborar y presentar los IPS según los plazos y
frecuencias descritos a continuación.
c) El titular del registro debe realizar un IPS para cada
producto que esté registrado y comercializado en Costa Rica.
d)
El titular del registro debe identificar la información de seguridad del producto
y la versión utilizada para la clasificación de las sospechas de RAM como
descritas o no descritas, e incluirlo como anexo en el IPS.
e)
El encargado de FV debe verificar los IPS y adjuntar una carta con su firma
para su presentación ante la DRPIS.
f)
El encargado de FV deberá informar al titular del registro para que éste
realice, si procede, como resultado de la evaluación beneficio/riesgo, las
modificaciones en la información de seguridad pertinentes en la monografía e
información para prescribir del expediente de registro sanitario, gestionándolo
a través de la Unidad de Registros y la plataforma Regístrelo.
12.8.2
La periodicidad de presentación está basada en la IBD y es la
siguiente:
|
Años transcurridos
(contados a partir del IBD)
|
Frecuencia
|
|
Primero
|
Cada 6 meses
|
|
Segundo
|
Cada 6 meses
|
|
Tercero
|
Cada año
|
|
Cuarto
|
Cada año
|
|
Quinto
|
Cada año
|
|
Sexto y posteriores
|
Cada 5 años
|
No
obstante el CNFV puede solicitar IPS fuera de estos periodos cuando por cuestiones
de riesgo, se determine la necesidad de su presentación.
12.8.3
El plazo para la entrega de los IPS contado desde la fecha de
cierre de datos (DLP, por sus siglas en inglés) y la presentación del informe
al CNFV, es el siguiente:
|
Intervalo de tiempo
cubierto por el IPS
|
Plazo
|
|
6 ó 12 meses
|
70 días naturales
|
|
Más de 12 meses
|
90 días naturales
|
NOTA: Para
facilitar el cumplimiento de los requisitos para los IPS, en cuanto a
contenido, el Ministerio de Salud mantendrá a disposición de los administrados
la Guía de presentación de IPS para la Industria Farmacéutica en su página web.
12.9
Planes de Minimización de los Riesgos:
12.9.1
Se debe contar con un plan estratégico de seguridad para
reducir los riesgos conocidos de los medicamentos en la etapa posterior a su
comercialización.
12.9.2
Los Planes de Minimización de Riesgos serán solicitados por
la DRPIS en caso de que sea requerido según normativa que expresamente los
exija, como en caso de medicamentos biológicos, o bien cuando por cuestiones de
riesgo, se determine la necesidad de su presentación.
12.10
Archivo:
El
sistema de gestión de archivo debe cumplir con los siguientes aspectos:
a)
Garantizar la conservación adecuada de la documentación relacionada con las
actividades de FV, así como su disponibilidad de una forma rápida y completa.
Las instalaciones del archivo pasivo o histórico deben ofrecer medidas de
protección de los materiales archivados contra posibles destrucciones por agua,
fuego, luz y plagas, entre otros.
b)
Las notificaciones de sospechas de RAM recibidas y cualquier documentación
adicional de seguimiento, así como los IPS y la correspondencia mantenida con
la DRPIS tienen que conservarse hasta al menos cinco años después de la finalización
de la comercialización del medicamento a que se refieren.
c)
El titular de registro debe conservar los PNT históricos por un período mínimo
de 10 años.
d)
Se debe conservar la documentación relativa al Currículum Vitae, entrenamiento
y formación del encargado de FV y del personal del departamento de FV, incluida
la de aquellos que ya no trabajan para el titular de registro, durante el
tiempo que el titular de registro mantenga su actividad y por 5 años más una
vez cesada dicha actividad.
e)
Se debe disponer de un sistema de registro de la documentación en el archivo
pasivo, con un sistema de control de entrada y salida de documentación del
mismo, en el que quede constancia de la documentación retirada, de la persona
que la retira y de la fecha de salida y retorno. Este sistema debe quedar
documentado en un PNT.
f)
El acceso al archivo pasivo o, en su caso, al archivo general, debe estar
restringido al personal autorizado.
g)
Si se produce un cambio de titularidad, el nuevo titular de registro debe tener
acceso a la información histórica de FV del medicamento en cuestión y
establecerse un acuerdo documentado entre ambos titulares. Cualquier
transferencia de materiales debe quedar documentada.
12.11
Auditorías:
El
titular de registro debe realizar auditorías periódicas del Sistema de
Farmacovigilancia, con el fin de comprobar que todas las actividades se
realizan de acuerdo a la legislación vigente y los PNT establecidos, para ello
debe:
a)
Establecer un programa de auditorías que especifique la frecuencia, el
contenido y el ámbito/alcance de las auditorías en función de la complejidad
del Sistema de Farmacovigilancia.
b)
Incluir en las auditorías todos los departamentos implicados en el Sistema de
Farmacovigilancia y sus respectivas actividades, ya sean del propio titular de
registro, de sus filiales, de cualquier empresa contratada y cualquier compañía
vinculada por acuerdos de FV.
c)
Designar a los responsables de llevar a cabo las auditorías. El personal
auditor, ya sea propio de la compañía o externo/contratado, debe ser
independiente del Sistema de Farmacovigilancia y estar debidamente calificado
por su formación y experiencia.
d)
Documentar en un informe, el resultado de cada auditoría que se remitirá a la
dirección del titular de registro y al encargado de FV. El titular de registro
debe registrar las auditorías realizadas y documentar las fechas de envío y de
recepción de los informes correspondientes.
e)
Establecer medidas correctivas para cada una de las deficiencias observadas y
se realizará un seguimiento documentado de su implementación.
f)
Mantener un archivo de las actividades de garantía de calidad, incluyendo
informes de auditoría, aplicación y seguimiento de medidas correctivas.
g) El
titular de registro no debe enviar a la DRPIS los informes de las auditorías
que realice, a menos que sean solicitados por la DRPIS con el fin de comprobar
las acciones correctivas tomadas.
h)
El procedimiento para realizar auditorías, la frecuencia de las mismas, así
como los aspectos a auditar deben estar establecidos en un PNT.
12.12
Acuerdos en materia de Farmacovigilancia:
12.12.1
El titular de registro puede subcontratar o transferir alguna
de las actividades derivadas de las obligaciones y responsabilidades en FV
señaladas en el numeral 12.1. Sin embargo el titular de registro es el
responsable final en materia de FV de los medicamentos de los cuales es
titular.
12.12.2
Debe existir un documento legal formalizado con terceros en
el caso de utilización de proveedores de servicios externos para la realización
de las actividades de Farmacovigilancia; el cual debe incluir una descripción
detallada de las actividades de FV asignadas a cada parte. Debe
quedar claro a cuál parte le corresponde intercambiar
información con el Ministerio de Salud. Las actividades no mencionadas en el
documento legal serán responsabilidad del titular de registro. Estos documentos
deben estar firmados y fechados por los representantes de ambas partes.
12.12.3
El titular de registro deberá asegurar que el encargado de FV
esté involucrado en la preparación o revisión de dichos acuerdos para verificar
que las responsabilidades de FV están cubiertas. Debe quedar documentado que el
encargado de FV conoce el contenido de dicho acuerdo. El titular del registro
debe establecer un mecanismo de revisión periódica y actualización de los
acuerdos existentes.
13.
COMISIÓN NACIONAL DE FARMACOVIGILANCIA.
13.1
La Comisión Nacional de Farmacovigilancia es un órgano de
asesoramiento que se encarga de evaluar las señales emitidas por el CNFV.
13.2
Obligaciones y responsabilidades:
a)
Analizar la información recibida del CNFV sobre señales detectadas a través de
los diferentes métodos de FV.
b)
Realizar la evaluación beneficio/riesgo de los medicamentos de acuerdo al PNT
establecido.
c)
Informar al CNFV acerca de los resultados de los análisis de las señales, para
la toma oportuna de medidas, cuando así se requiera.
d)
Todas las actividades que realicen la Comisión Nacional de Farmacovigilancia,
se harán de acuerdo a lo establecido en el PNT de la Comisión Nacional de
Farmacovigilancia.
14.
AUTORIDAD REGULADORA
14.1
CENTRO NACIONAL DE FARMACOVIGILANCIA.
Además de las establecidas
en el Decreto Ejecutivo N° 35244-S "Reglamento del Sistema Nacional de
Farmacovigilancia", son obligaciones y responsabilidades del CNFV:
14.1.1
Clasificación del riesgo detectado en:
a)
Riesgo inminente y grave para la salud
b)
Riesgo aceptable en todas las condiciones de uso
c)
Riesgo solo aceptable en determinadas condiciones de uso
d)
Riesgo inaceptable en todas las condiciones de uso.
14.1.2
Manejo de la crisis:
Analizar
la información disponible y definir en función de ésta, las decisiones pertinentes
que incluyen la aplicación de las medidas sanitarias, la búsqueda o generación
de información adicional, así como la comunicación de la situación de riesgo si
lo hubiere o la inexistencia del mismo.
En
cualquier caso, debe establecerse una cooperación estrecha entre las partes
involucradas y la DRPIS podrá tomar medidas urgentes cuando se tengan pruebas
del riesgo y del impacto sobre la salud pública.
14.2
DIRECCIÓN DE REGULACIÓN DE PRODUCTOS DE INTERÉS SANITARIO
Para
efectos del presente decreto le corresponde a la DRPIS ejecutar las siguientes
funciones:
14.2.1
Aplicación de medidas administrativas para reducir el riesgo:
La
DRPIS debe aplicar las medidas administrativas necesarias para mantener la
relación beneficio/riesgo favorable de los medicamentos que se comercializan en
el país.
14.2.2
Comunicación de los riesgos asociados a los medicamentos:
Toda
nueva información de seguridad debe comunicarse tanto a los profesionales en
salud como a los titulares del registro, los sistemas de vigilancia
establecidos u otras instituciones.
Se
difundirán las medidas adoptadas utilizando los canales de comunicación
apropiados, entre ellas:
a) El etiquetado oficial establecido.
b)
Carta de respuesta a quejas y reclamaciones.
c)
Comunicaciones de riesgo dirigidas a profesionales de la salud.
d)
Resoluciones de medidas sanitarias de reducción de riesgos.
e)
Boletines disponibles de forma impresa, distribuidos por correo electrónico o
en Internet.
f)
Artículos científicos
g)
Advertencias públicas en medios de difusión masiva (prensa escrita, radio,
televisión o Internet).
14.2.3
Proveer al CNFV los recursos necesarios para el buen
funcionamiento y ejecución de las actividades de FV.
14.2.4
Elaborar la legislación o la reglamentación necesaria para
mantener la relación beneficio/riesgo favorable de los medicamentos que se
comercializan en el país.
15.
ESTUDIOS POST-COMERCIALIZACIÓN.
Toda
sospecha de RAM que se presente durante la realización de estudios de
medicamentos en fase 4 o fase postcomercialización, se deben notificar al CNFV
de acuerdo a los plazos y mecanismos establecidos en el Decreto Ejecutivo N°
35244 - S, "Reglamento del Sistema Nacional de Farmacovigilancia."
16.
BIBLIOGRAFÍA.
16.1 España.
Buenas Prácticas de Farmacovigilancia para la Industria Farmacéutica
medicamentos de uso humano, 21 de diciembre 2011.
16.2 España.
Buenas Prácticas de Farmacovigilancia del Sistema Español de Farmacovigilancia
de medicamentos de uso humano, 17 de setiembre 2002.
16.3 México.
Norma Oficial Mexicana NOM-220-SSA1-2002, instalación y operación de la
Farmacovigilancia. Diario Oficial de la Federación, 27 de julio 2004.
16.4 ORGANIZACIÓN
PANAMERICANA DE LA SALUD. Buenas Prácticas de Farmacovigilancia para las Américas.
Washington, DC.: OPS, © 2011. (Red PARF Documento Técnico No. 5).
16.5 Paraguay. Manual de Reglamentos, Procedimientos
y Buenas Prácticas de Farmacovigilancia del Paraguay, junio 2011.