N° 44917 - S
EL
PRESIDENTE DE LA REPÚBLICA
Y LA
MINISTRA DE SALUD
En
uso de las facultades que les confiere los artículos 140 incisos 3) y 18) y 146
de la Constitución Política; 25 inciso 1 ), 27 inciso 1 ), 28 inciso 2) acápite
b) y 103 inciso 1) de la Ley No.6227 del 02 de mayo de 1978 "Ley General
de la Administración Pública"; 1, 2, 4, 7, y 49 de la Ley No. 5395 del 30
de octubre de 1973 "Ley General de Salud", 1, 2 y 6 de la Ley No.
5412 del 8 de noviembre de 1973 "Ley Orgánica del Ministerio de
Salud"; 1 al 6 de la Ley No. 8220 del 04 de marzo de 2002 "Protección
al ciudadano del exceso de requisitos y trámites administrativos"; 4 de la
Ley No. 10113 del 2 de marzo de 2022 "Ley del Cannabis para uso Medicinal
y Terapéutico y del cáñamo para uso alimentario e industrial"; 39, 41 y 49
de la Ley No. 10473 del 24 de abril de 2024 "Sistema Nacional para la
Calidad"; 1 de la Ley No. 7472 del 20 de diciembre del 1994 "Ley de
Promoción de la Competencia y Defensa Efectiva del Consumidor" y 7 y Anexo
B de la Ley No. 7475 del 20 de diciembre de 1994 "Acta Final en que se
incorporan los Resultados de la Ronda Uruguay de Negociaciones Comerciales
Multilaterales y Crea Organización Mundial del Comercio".
CONSIDERANDO:
1.-Que
es un deber ineludible del Estado velar por la salud de la población, evitando
o reprimiendo aquellos actos u omisiones de particulares que impliquen un
riesgo para la salud humana que es un bien jurídico de importancia suprema para
el desarrollo social y económico del país.
2.-Que
el artículo 1 de la Ley No. 10113 del 2 de marzo del 2022 "Ley del
cannabis para uso medicinal y terapéutico y del cáñamo para uso alimentario e
industriar', señala como uno de los objetivos de la ley, el de regular y
permitir el acceso y la utilización del cannabis y sus derivados exclusivamente
para uso medicinal y terapéutico, a fin de garantizar el derecho fundamental a
la salud de toda la población costarricense.
3
.-Que el artículo 26 de la Ley No. 10113 del 2 de marzo del 2022 "Ley
del cannabis para uso medicinal y terapéutico y del cáñamo para uso alimentario
e industrial", establece que el Ministerio de Salud determinará las
medidas pertinentes para lograr el control y la vigilancia del cumplimiento de
las obligaciones de las personas productoras, licenciatarias y permisionarias.
Para ello, podrá regular y ejercer actividades de control sobre las etapas de
producción, transporte, importación, exportación, transformación y distribución
de los productos regulados por la Ley No. 10113 del 2 de marzo del 2022 "Ley
del cannabis para uso medicinal y terapéutico y del cáñamo para uso alimentario
e industrial".
4.-Que
como consecuencia de una revisión efectuada por el Comité de Expertos en
Farmacodependencia de la Organización Mundial de la Salud (OMS) y aprobada por
la Comisión de Estupefacientes (CND) de Naciones Unidas, se retiraron las
sustancias "cannabis y resina de cannabis" de la Lista IV de la
Convención Única sobre Estupefacientes de 1961 enmendada por el Protocolo de
1972 -reconociendo su potencial ventaja terapéutica.
5
.-Que el uso del cannabis presenta un importante potencial terapéutico en
diversas patologías, y la industria asociada a su producción crece a nivel
nacional e internacional, y que por sus características puede contribuir a la
creación de empleo y al desarrollo industrial del país y su posicionamiento en
la región.
6.-Que
existen experiencias a nivel internacional que evidencian que, en un marco de
seguridad y calidad, junto con el acompañamiento médico adecuado, se reducen
los daños potenciales que el uso del cannabis puede producir en un mercado
deficiente de controles.
7.-Que
la conformación de una nueva categoría de productos medicinales a base de
cannabis, que cumplan los requisitos establecidos de regulación y control tanto
de los insumos utilizados en los procesos productivos como del producto
elaborado, es de singular relevancia para dar cumplimiento a los objetivos
planteados en la Ley 10113 "Ley del cannabis para uso medicinal y
terapéutico y del cáñamo para uso alimentario e industrial".
8.-Que
permitir la elaboración y comercialización de productos medicinales a base de
cannabis, bajo una categoría diferente a la de medicamento o productos
naturales medicinales, proporcionará mayor acceso y disponibilidad de productos
pertenecientes a la categoría establecida, pudiendo considerar además su
aplicación en diversas patologías.
9.-Que
los productos medicinales a base de cáñamo y/o cannabis psicoactivo que
conformen la categoría establecida estarán disponibles en farmacias para su
dispensación bajo receta médica y, por lo tanto, es necesario que los médicos
tratantes tengan garantías de la calidad de los productos que prescriben, que
redundarán en garantizar la seguridad de los pacientes.
10.-Que
por lo tanto resulta necesario y conveniente elaborar la reglamentación
aplicable a los efectos de establecer un marco regulatorio eficaz y ágil,
favoreciendo el desarrollo de la producción de productos medicinales a base de
cannabis, previendo asimismo los mecanismos para que las autoridades con
competencia en la materia ejerzan controles adecuados para la protección de la
salud y la seguridad de la población;
11.-
Que en concordancia con lo establecido el artículo 361 de la Ley No. 6227 del 2
de mayo de 1978 "Ley General de la Administración Pública" el
presente Decreto Ejecutivo cumplió con el trámite de consulta pública, la misma
se realizó en la plataforma del Ministerio de Economía Industria y Comercio,
denominada Sistema de Control Previo, SICOPRE, en fecha 07 de octubre al 18 de
octubre del 2024, siendo que fueron atendidas todas las observaciones emitidas
por los administrados.
12.-
Que la propuesta de reglamentación cuenta con criterio técnico vinculante
positivo No. DCAL-CONART-OF-002-2024 del Consejo Nacional de Reglamentación
Técnica (CONART), asimismo se realizó la consulta internacional correspondiente
ante la Organización Mundial del Comercio, OMC, en fecha 02 de diciembre del
2024 al 02 de febrero del 2025, de dicha consulta se recibieron observaciones
las cuales fueron atendidas.
13.-
Que de conformidad con lo establecido en el artículo 12 bis del Decreto
Ejecutivo No. 37045-MP-MEIC de 22 de febrero de 2012 "Reglamento a la Ley
de Protección al Ciudadano del Exceso de Requisitos y Trámites Admin istrativos"
y su reforma, esta regulación cumple con los principios de mejora regulatoria,
de acuerdo con el informe Nº DMR-DARINF-253-2024, emitido por la Dirección de
Análisis Regulatorio de la Dirección de Mejora Regulatoria del Ministerio de
Economía, Industria y Comercio.
POR TANTO,
DECRETAN:
Artículo 1. Aprobar el siguiente reglamento técnico:
"REGLAMENTO
TÉCNICO RTCR 515:2024. CANNABIS. PRODUCTOS
MEDICINALES
A BASE DE CANNABIS. DISPOSICIONES ADMINISTRATIVAS,
REGISTRO
SANITARIO, ETIQUETADO, ESPECIFICACIONES,
CONTROL Y
PUBLICIDAD"
1.- OBJETO. El presente reglamento tiene por objeto,
establecer las disposiciones administrativas y requisitos para regular y
controlar los productos medicinales a base de cannabis.
2.- ÁMBITO DE APLICACIÓN. El presente reglamento
aplica a los productos industrializados, en adelante denominados productos
medicinales a base de cannabis y materias primas que contienen derivados de la
planta de cannabis, destinados al uso y aplicación en la medicina humana.
3.- REFERENCIAS. Este reglamento se complementa
con la siguiente legislación:
3.1.
Decreto Ejecutivo Nº 44585-MP-MAG-MS del 12 de julio del 2024 "Reglamento
a la Ley Nº 10113, Ley del Cannabis para uso medicinal y terapéutico y del
Cáñamo para uso alimentario e industrial del 02 de marzo del 2022, Reglamento
del Cáñamo para Uso Alimentario e Industrial o Uso Medicinal o
Terapéutico", publicado en Alcance No. 140 a la Gaceta Nº 149 del 14 de
agosto del 2024.
3.2.
Decreto Ejecutivo Nº 44695- MP- MAG-S del 29 de agosto del 2024
"Reglamento a la Ley Nº 10113 del 02 de marzo del 2022 "Ley del
Cannabis para uso medicinal y terapéutico y del Cáñamo para uso alimentario e
industrial del 02 de marzo del 2022, Reglamento del Cannabis para Uso Medicinal
y Terapéutico", publicado en Alcance No. 173 a La Gaceta No. 195 del 18 de
octubre del 2024.
3.3.
Decreto Ejecutivo Nº 31969-S del 26 de mayo del 2004 "Manual de Normas
para la Habilitación de Farmacias", publicado en La Gaceta Nº 175 del 07
de setiembre del 2004.
3.4.
Decreto Ejecutivo Nº 37111-S del 12 de enero del 2012 "Reglamento para el
Control de Drogas Estupefacientes y Psicotrópicos", publicado en el
Alcance Nº 72 a la Gaceta Nº 108 del 05 de junio del 2012.
3.5.
Decreto Ejecutivo Nº 43259-COMEX- S-MEIC del 27 de setiembre del 2021
"Publicación de la resolución Nº 446-2021 (COMIECO-XCIV) de fecha 28 de
abril del 2021 y sus Anexos: "Anexo I: Reglamento Técnico Centroamericano
RTCA 11.03.59:18 Productos Farmacéuticos. Medicamentos para Uso Humano. Requisitos
de Registro, Sanitario" y Anexo II: Reconocimiento Mutuo de Registro
Sanitario de Medicamentos
para Uso
Humano", publicado en el Alcance Nº 245 a la Gaceta Nº 232 del 02 de
diciembre del 2021.
3.6.
Decreto Ejecutivo Nº 36638-COMEX-S-MEIC del 30 de marzo del 2011 "Publicación
de la resolución Nº 256-2010 (COMIECO-LIX) de fecha 13 de diciembre de 2010:
Reglamento Técnico Centroamericano RTCA 11.01.04:10 Productos Farmacéuticos.
Estudios de Estabilidad de Medicamentos para Uso Humano", publicado en la
Gaceta Nº 129 del 05 de julio del 2011.
3.7.
Decreto Ejecutivo Nº 43432-S del 09 de marzo del 2022 "Reglamento general
para permisos sanitarios de funcionamiento, permisos de habilitación y
autorizaciones para eventos temporales de concentración masiva de personas,
otorgados por el Ministerio de Salud", publicado en el Alcance Nº 60 a la
Gaceta Nº 56 del 23 de marzo del 2022.
3.8.
Decreto Ejecutivo Nº 44435-COMEX-MEIC-S del 09 de octubre del 2023 Publicación
de la Resolución Nº 472-2023 (COMIECO-EX) de fecha 05 de junio de 2023: Aprueba
el "Reglamento Técnico Centroamericano RTCA 11.03.64:19 Productos
Farmacéuticos. Productos Naturales Medicinales Para Uso Humano. Requisitos de
Registro Sanitario" y el "Reconocimiento Mutuo de Registro Sanitario
de Productos Naturales Medicinales para uso Humano"; y deroga la
Resolución Nº 303-2013 (COMIECO-EX) del 15 de mayo de 2013 promulgada mediante
el Decreto Ejecutivo Nº 37851-COMEX-MEIC-S de fecha 27 de mayo de 2013,
publicado en el Alcance Nº 95 a la Gaceta Nº 89 del 20 de mayo del 2024.
3.9.
Decreto Ejecutivo Nº 42918-COMEX-MEIC-S del 18 de enero del 2021 Publicación de
la resolución Nº 423-2020 (COMIECO-XC) fecha de 30 de abril del 2020 y sus
Anexos: "Reglamento Técnico Centroamericano RTCA 11.03.69:13
Productos Farmacéuticos. Productos Naturales Medicinales para uso humano.
Buenas Prácticas de Manufactura y su Guía de Verificación", publicado en
el Alcance Nº 75 a la Gaceta Nº 73 del 16 de abril del 2021.
3.10.
Decreto Ejecutivo Nº 37988-S del 03 de octubre del 2013 "Reglamento para
el Funcionamiento y la Utilización del Portal "Regístrelo", publicado
en la Gaceta Nº 203 del 22 de octubre del 2013.
3.11.
Decreto Ejecutivo N º 39984-S del 01 de setiembre del 2016 "Reglamento de
Utilización y Funcionamiento del Sistema Automatizado de Receta Digital de
Psicotrópicos y Estupefacientes", publicado en el Alcance Nº 252 en la
Gaceta N º 215 del 09 de noviembre del 2016.
3.12.
Decreto Ejecutivo Nº 36868-S del 12 de setiembre del 2011 "Reglamento para
la autorización y control sanitario de la publicidad de productos de interés
sanitario", publicado en la Gaceta Nº 236 del 08 de diciembre del 2011.
4.- DEFINICIONES. Para los efectos de
interpretación del presente reglamento se establecen las siguientes
definiciones:
4.1. Acreditación: atestación de tercera parte
relativa a un organismo de evaluación de la conformidad que manifiesta la
demostración formal de su competencia, su imparcialidad y su operación
coherente al llevar a cabo actividades específicas de evaluación de la
conformidad, según las normas especificadas.
4.2. Alcance de la acreditación: actividades
específicas de evaluación de la conformidad para las que se pretende o se ha
otorgado la acreditación.
4.3. Autoridad competente: autoridad responsable de
la emisión del Certificado de Libre Venta y Certificado de Buenas Prácticas de
Manufactura para productos medicinales a base de cannabis o equivalentes en
cada país o región.
4.4. Autorización: acto administrativo en virtud del
cual el MAG o el MS autoriza a una persona física o jurídica a desarrollar las
actividades establecidas en el Decreto Ejecutivo Nº 44585-MP-MAG-S del 12 de
julio del 2024 Reglamento a la Ley Nº I0l 13, "Ley del Cannabis para uso
medicinal y terapéutico y del Cáñamo para uso alimentario e industrial del 02 de
marzo del 2022", Reglamento del Cáñamo para Uso Alimentario e Industrial o
Uso Medicinal o Terapéutico.
4.5. Buenas Prácticas de Manufactura: conjunto
de procedimientos y normas destinados a garantizar la producción uniforme de
los lotes de productos medicinales a base de cannabis para uso humano, que
satisfagan las normas de calidad.
4.6. Cadena de custodia: registro que documenta
completamente la posesión de muestras y cualquiera transferencia desde el
momento de la recolección hasta la recepción en el laboratorio hasta su
disposición final.
4.7. Calidad: naturaleza esencial de un
producto y todos sus atributos y propiedades, que determinan su idoneidad a los
propósitos a los que se destina.
4.8. Cannabinoides: metabolitos secundarios
producidos por las plantas del género cannabis de tipo terpeno fenólicos, que
son asociados con la actividad farmacológica que presenta el cannabis.
4.9. Cannabinoide sintético: cannabinoides obtenidos
por síntesis química o por la modificación sintética del metabolito secundario
extraído de la planta que interactúan con los receptores cannabinoides para
mimetizar acciones análogas a los cannabinoides naturales.
4.10. Cannabis: toda planta herbácea del género
cannabis (familia Cannabaceae), incluyendo sus semillas, hojas,
sumidades floridas o con fruto y cualquier otro material vegetal proveniente de
esta.
4.11. CBD o Cannabidiol: cannabinoide no psicoactivo con
aplicaciones alimentarias, industriales, investigativas, terapéuticas y
médicas, que contiene la planta del cannabis.
4.12. Certificado de Buenas Prácticas de Manufactura: documento
expedido por la autoridad competente en el país en el cual se encuentra ubicado
el laboratorio fabricante, donde se certifica que el laboratorio cumple
con las Buenas Prácticas de Manufactura o su equivalente.
4.13.
Certificado de Libre Venta: documento expedido por la autoridad competente del
país de origen o de procedencia, donde se certifica que el producto o productos
a que se refiere el certificad.o están autorizados para la venta, uso o
distribución en el país origen o de procedencia. En caso de fabricación por
terceros o filiales y que el producto no se comercialice en el país de
fabricación, el certificado podrá ser emitido por la autoridad competente del
país del titular del producto.
4.14. Certificado o informe de análisis: informe
emitido por un laboratorio de ensayo que describe los resultados de las pruebas
analíticas realizadas a un producto.
4.15. Contrato de fabricación: documento legal celebrado
entre el titular del producto medicinal a base de cannabis y el fabricante en
el cual se establecen las condiciones, compromisos y demás circunstancias para
la fabricación de uno o más productos.
4.16. Cosecha: proceso de obtención y/o
recolección del material vegetal proveniente de la planta de cannabis, durante
o después del ciclo de cultivo según sea la finalidad de este.
4.17.
Demostración de la conformidad por· primera parte: modalidad de
demostración basada en una evaluación realizada por el fabricante o importador
y materializada en un documento formalmente emitido.
4.18. Derivados de cannabis: Compuestos o ingredientes
que se extraen del cannabis y que está destinado a ser utilizado en o para un
producto medicinal a base de cannabis de dosificación. Pueden ser derivados de
cannabis no psicoactivo o derivad.os de cannabis psicoactivo.
4.19. Derivados no psicoactivos de cannabis: aceites,
resinas, tinturas y extractos crudos, purificados o procesados, otros productos
o innovaciones resultado del desarrollo tecnológico obtenidos a partir del
cannabis y/o del componente vegetal cuyo contenido de tetrahidrocannabinol
(THC) incluyendo sus isómeros, sales y formas ácidas es inferior al uno por
ciento (1 %), los cuales serán usados para la fabricación de productos
medicinales a base de cannabis, sin ser producto terminado.
4.20. Derivados psicoactivos de cannabis: aceites, resinas,
tintnras, extractos crndos, purificados o procesados, otros
productos o innovaciones resultado del desarrollo tecnológico obtenidos a
partir del cannabis y/o del componente vegetal cuyo contenido de
tetrahidrocannabinol (THC) incluyendo sus isómeros, sales y formas ácidas que
igual o supera el uno por ciento (1 %) los cuales serán usados para la
fabricación de productos medicinales a base de cannabis, sin ser producto
terminado.
4.21. Dispensación: acto profesional del farmacéutico
de proporcionar uno o más productos medicinales a base de cannabis a un
paciente, en atención a la presentación de una receta médica elaborada por un
profesional autorizado. En este acto el farmacéutico informa y orienta al
paciente sobre el uso adecuado de los productos, reacciones adversas,
interacciones medicamentosas y las condiciones de conservación del producto.
4.22. Empaque o envase primario: recipiente
dentro del cual se coloca directamente el producto medicinal a base de cannabis
en la forma terminada.
4.23. Empaque o envase secundario: envase
definitivo de distribución y comercialización o material de empaque en el que
se coloca el envase primario que contiene el producto.
4.24. Especificación: descripción de los requisitos
(criterios de aceptación para los procedimientos de ensayo establecidos) con
los que la sustancia o producto tiene que cumplir para asegurar una calidad
adecuada.
4.25. Estudios de estabilidad: pruebas
que se efectúan para obtener información sobre las condiciones en las que se
deben procesar y almacenar las materias primas o los productos semielaborados o
los productos terminados, según sea el caso. Las pruebas de estabilidad también
se emplean para determinar periodo de validez del producto medicinal a base de
cannabis en su envase primario original y en condiciones de almacenamiento
especificadas
4.26. Etiqueta para material vegetal o derivados: cualquier
marbete, rótulo, marca, imagen u otra materia descriptiva o gráfica, que se
haya escrito, impreso, estarcido, marcado, marcado en relieve o en huecograbado
o adherido al envase o empaque, que identifica y describe el material vegetal o
derivado contenido en él.
4.27. Etiqueta complemeutaria para material vegetal o derivados:
aquella que se utiliza para colocar la información obligatoria
cuando en la etiqueta original esta se encuentra en un idioma diferente al
español o para agregar aquellos elementos obligatorios no incluidos en la
etiqueta original y que el presente reglamento exige, en el caso del empaque
del material vegetal o derivados.
4.28. Etiquetado de producto medicinal a base de cannabis: información
obligatoria incluida en la etiqueta, rótulo imagen u otra materia descriptiva o
gráfica que se haya escrito, impreso, estarcido o marcado en relieve que se
adhiere o incluye en el envase de un producto medicinal a base de cannabis.
4.29. Etiquetado para material vegetal o derivados: es la
información obligatoria incluida en la etiqueta, rótulo, imagen u otra materia
descriptiva o gráfica que se haya escrito, impreso, estarcido, marcado en
relieve, que se adhiere o incluye en el envase o empaque que contiene material
vegetal o un derivado de cannabis.
4.30. Evaluación de la conformidad: demostración
de que se cumple los requisitos especificados en los reglamentos técnicos o
normas relativos a un producto, proceso, sistema, persona u organismo.
4.31. Examen previo aduanal: inspección o
reconocimiento de mercancías bajo la supervisión aduanera, efectuado por el
consignatario o el agente aduanero que lo representa, con el propósito de
declarar correctamente la información o los datos exigibles para el despacho de
las mercancías.
4.32. Excipiente: sustancia sin acción
farmacológica a la concentración utilizada que determina o modifica la
consistencia, forma, o propiedades fisicoquímicas de sus preparaciones de
productos medicinales a base de cannabis.
4.33.
Fabricación: todas las operaciones involucradas en la compra de materiales y
productos, producción, acondicionamiento, control de calidad, aprobación,
almacenamiento, distribución del producto terminado y los controles
relacionados.
4.34. Fabricación a terceros: fabricación nacional o
extranjera realizada dentro de los límites de una contratación previa entre el
titular del producto medicinal a base de cannabis y el fabricante.
4.35. Fabricación de derivados: proceso
de transformación del cannabis y/o del material vegetal en derivados.
4.36. Fecha de expiración o vencimiento: fecha
establecida para cada lote colocada en el empaque primario y secundario hasta
la cual se espera que el producto medicinal a base de cannabis, almacenado
adecuadamente cumpla las especificaciones de calidad.
4.37. Fines medicinales: uso de los productos medicinales
a base de cannabis como terapia complementaria para tratar algunas enfermedades
o aliviar determinados síntomas.
4.38. Forma farmacéutica: es la forma física que se
le da a un producto medicinal a base de cannabis, para facilitar la
administración del producto al paciente.
4.39. INTE-1SO/IEC 17025: norma internacional que
establece los requisitos generales para la competencia en la realización de
ensayos o de calibraciones, incluido el muestreo. Contempla los ensayos y las
calibraciones que se realizan utilizando métodos normalizados, métodos no
normalizados, métodos normalizados modificados y métodos desarrollados por el
propio laboratorio. Es aplicable a todas las organizaciones que realizan
ensayos o calibraciones y los laboratorios en los que los ensayos o las
calibraciones forman parte de la inspección y la certificación de productos.
4.40. Laboratorio de ensayo: sitio en el cual se
realizan operaciones/ensayos y/o muestreo para identificar y/o medir las características
de un producto, proceso o servicio determinado con un método específico.
4.41.
Licencia: acto administrativo mediante el cual el MAG o el MS, habilitan a una
persona física o jurídica a desarrollar las actividades establecidas en el
Decreto Ejecutivo N º 44695-MP-S-MAG del 29 de agosto del 2024 Reglamento a la
Ley Nº 10113 del 02 de marzo del 2022 "Ley del Cannabis para uso medicinal
y terapéutico y del Cáñamo para uso alimentario e industrial" Reglamento
del Cannabis para Uso Medicinal y Terapéutico.
4.42. Límite de cuantificación: concentración
mínima de analito en la matriz de una muestra que puede ser cuantificada con
una exactitud y precisión aceptable bajo condiciones analíticas específicas.
Los límites de cuantificación son característicos de desempeño que marcan la
habilidad de un proceso de medición química para cuantificar adecuadamente un
analito.
4.43. Límite de detección: concentración mínima de un
analito en la matriz de una muestra que puede ser detectada, pero no
necesariamente cuantificada, bajo condiciones analíticas específicas.
4.44. Lista de empaque: es un documento que detalla el
contenido específico de cada paquete y es complemento de la factura comercial,
ya que suministra los detalles fisicos de la carga, tales como: descripción,
cantidad de productos, cantidad de cajas, peso y tamaño.
4.45. Lote: cantidad de producto medicinal a base de
cannabis para uso humano terminado que se produce en un ciclo de producción. La
característica esencial del lote es su homogeneidad.
4.46. Material de referencia certificado: material
de referencia, caracterizado por un procedimiento válido desde el punto de
vista metrológico para una o más de las propiedades especificadas, acompañado
por un certificado que proporciona el valor de la propiedad especificada, su
incertidumbre asociada y una declaración de la trazabilidad metrológica.
4.47. Material de referencia: material homogéneo y
estable respecto a una o más de las propiedades especificadas, apropiado para
el uso para el que se destina en un proceso de medición.
4.48. Material vegetal: se refiere a la planta de
cannabis y cualquier parte de esta que puede usarse como materia prima para la
fabricación de derivados de cannabis o productos medicinales a base de
cannabis; se obtiene de la planta en cualquier momento de su ciclo de vida y
producto de la cosecha.
4.49. Método de ensayo: documento que describe un
conjunto de operaciones, descritas específicamente, para realizar mediciones
particulares, respecto a las características de un ítem.
4.50. Método desarrollado por el laboratorio: método de
medición que no se encuentra en normas u otras colecciones de métodos, ni en
publicaciones de terceros, habiendo sido desarrollado por el propio
laboratorio. El desarrollo del método, así como su adaptación, incluyen la
etapa de su validación
4.51. Método no normalizado: método desarrollado por un
tercero ( organismo no reconocido a nivel internacional), o que ha sido
adaptado por el laboratorio a partir de un método normalizado. El desarrollo
del método, así como su adaptación, incluyen la etapa de su validación.
4.52. Método normalizado: método desarrollado por un
organismo de normalización u otro organismo reconocido a nivel internacional,
cuyos métodos son generalmente aceptados por el sector técnico correspondiente.
4.53. Método normalizado modificado: método
normalizado al cual, por diversas razones, se le realiza una o varias
modificaciones que pudiesen influenciar de manera significativa los resultados
obtenidos. Cualquier cambio a un método normalizado debe analizarse y demostrar
técnicamente su efecto sobre los resultados obtenidos, esto debe estar
debidamente documentado.
4.54. Modificaciones o cambios post-registro: modificaciones
al registro sanitario de un producto medicinal a base de cannabis posterior al
otorgamiento de su registro.
4.55. Muestra: parte o porción finita extraída
de un conjunto que se considera representativa del total y que se toma o se
separa de ella con ciertos métodos para someterla a estudio, análisis o
experimentación.
4.56. Muestra sin valor comercial: producto
que no será comercializado y que puede ser utilizado para ensayos o pruebas con
fines de investigación, estudios de mercado o cualquier otro siempre que no sea
para comercializarse en el territorio nacional.
4.57. Notificación: acto administrativo por el que se
comunica al interesado que el material vegetal derivado de cannabis a emplear
como materia prima para elaborar productos medicinales a base de cannabis ha
cumplido o no con los requisitos de este reglamento y puede utilizarse o no
para ese fin.
4.58. Organismo de Evaluación de la Conformidad: organismo
que realiza servicios de evaluación de la conformidad, pueden ser de naturaleza
pública o privada, nacionales o extranjeros.
4.59. País de origen: país donde se realiza el proceso
de fabricación del producto medicinal a base de cannabis.
4.60. Preparación magistral: producto medicinal
elaborado por el regente farmacéutico en una farmacia que cuenta con licencia o
autorización para fabricación de preparaciones magistrales a base de cannabis,
para atender una prescripción o receta médica de un paciente individual.
4.61. Prescripción: acto realizado por un profesional
de la salud autorizado, que consiste en la emisión de una orden suscrita para
dispensar uno o más productos medicinales a base de cannabis especificados en
ella. La prescripción debe ser respaldada mediante la emisión de una receta médica.
4.62. Postcosecha: conjunto de actividades que
inicia desde la recolección de la cosecha, hasta la culminación de las
diferentes prácticas de acondicionamiento para su posterior procesamiento.
4.63. Producto medicinal a base cannabis: producto
seco (flores e inflorescencia de cannabis) o un producto de dosificación,
procesados, empacados y etiquetados, que poseen una concentración definida de
CBD y/o THC y que se utiliza para fines medicinales. Un producto medicinal a
base de cannabis no contiene sustancias activas ni cannabinoides de origen
sintéticos.
4.64. Producto de dosificación: producto
que contiene una concentración establecida que permite que se pueda administrar
en una cantidad y tiempo definidos.
4.65. Productos secos de cannabis: material
vegetal (flores e inflorescencias) que ha tenido un proceso de secado para
alcanzar un límite de humedad especifico.
4.66. Profesional responsable: profesional
farmacéutico responsable del trámite de registro sanitario ante el MS,
autorizado por el titular del producto o su representante legal a través de un
poder otorgado de acuerdo con la legislación vigente.
4.67. Publicidad: cualquier forma de mensaje que
sea difundido, de cualquier modo, en el ejercicio de una actividad comercial,
industrial, artesanal o profesional con el objeto de promover la venta de
productos, bienes muebles, inmuebles, la constitución o la transferencia de
derechos y obligaciones, o bien la prestación de servicios, así como la
difusión de ideas determinadas.
4.68. Receta médica: documento físico o digital que
contiene la prescripción médica emitida y suscrita por un profesional de la
salud autorizado. La receta médica se requiere para la dispensación de
productos medicinales a base de cannabis.
4.69. Receta digital: formulario electrónico
oficializado en el sistema electrónico, que contiene la orden extendida por los
profesionales legalmente autorizados para ello, en que se prescribe al paciente
el producto medicinal a base de cannabis en ella indicado.
4.70.
Registro sanitario: aprobación por parte del MS para la comercialización de un
producto medicinal a base de cannabis, después de la evaluación de la
documentación requerida.
4.71.
Representante legal: persona física o jurídica con domicilio en Costa Rica,
autorizada por el titular del producto medicinal a base de cannabis mediante un
poder legalizado o apostillado, que responde legalmente ante el MS.
4.72. Titular del producto o titular del registro: persona
física o jurídica propietaria del producto.
4.73. THC o tetrahidrocannabinol: es el
componente psicoactivo (alteración de la percepción y modificación del estado
de ánimo) de la planta de cannabis más importante y abundante en las variedades
clasificadas precisamente como psicoactivas.
4.74. Unidad de dosis producto medicinal a base de cannabis: forma de
presentación de un producto medicinal a base de cannabis con la cantidad de
cannabinoides, ya sea CBD y/o THC necesaria para administrar una sola dosis.
4.75. Uso personal: se refiere al ingreso al país de
productos medicinales a base de cannabis destinados exclusivamente a ser
utilizados o consumidos por la persona física que gestione el trámite de
ingreso.
4.76. Validación: acción documentada que demuestra
que un procedimiento, proceso, equipo, material, actividad o sistema conducen a
los resultados previstos.
5.- SÍMBOLOS Y ABREVIATURAS. Para los efectos de este
Reglamento se entenderá por:
5.1. AOAC: Asociación de Colaboración Analítica Oficial
(Association of Official Analytical Collaboration).
5.2. ASTM: Sociedad Estadounidense para Pruebas y
Materiales (American Society far Testing and Materials).
5.3. BPM: Buenas Prácticas de Manufactura.
5.4. CBDA: Ácido cannabidiólico.
5.5. DRPIS: Dirección de Regulación de Productos de
Interés Sanitario.
5.6. ECA: Ente Costarricense de Acreditación.
5.7 FAD: Formulario Aduanero de Desalmacenaje.
5.8. INTECO: Instituto de Normas Técnicas de Costa Rica.
5.9. ISO: Organización Internacional de Normalización
(Intemational Organization far Standardization).
5.10. MAG: Ministerio de Agricultura y Ganadería.
5.11. MS: Ministerio de Salud.
5.12.
PSF: Permiso Sanitario de Funcionamiento.
5.13. PROCOMER: Promotora de Comercio Exterior.
5.14. RTCA: Reglamento Técnico Centroamericano.
5.15. THCA: ácido tetrahidrocannabinólico.
5.16. UR: Unidad de Registros
5.17. USP: Farmacopea de los Estados Unidos.
5.18. VUCE: Sistema de Ventanilla Única de Comercio
Exterior.
5.19. A9-THCA: ácido delta-9-tetrahidrocannabinólico.
5.20. A9-THC: delta-9-tetrahidrocannabinol.
6.- DISPOSICIONES GENERALES DE LOS PRODUCTOS MEDICINALES A BASE DE
CANNABIS. Se establecen las siguientes disposiciones generales para los
productos medicinales a base de Cannabis:
6.1. Productos autorizados. Se autoriza la fabricación
e importación de los siguientes productos medicinales a base de cannabis:
6.1.1.
Productos secos de cannabis: flores e inflorescencias secas y empacadas.
6.1.2.
Productos de dosificación a base de cannabis en una forma farmacéutica
determinada ( como tabletas, cápsulas, cremas o un líquido oral), el cual puede
contener excipientes. Se excluyen las formas farmacéuticas estériles.
6.1.3.
Preparaciones magistrales. Deben ser elaboradas a partir de derivados de
cannabis y solo se acepta la preparación de formas farmacéuticas no estériles.
Las preparaciones magistrales a base cannabis no requieren registro sanitario.
6.2.
Vías de administración. La vía de administración de productos secos de cannabis
debe ser por vía oral por medio de infusión o inhalada, el fumar no es un
método recomendado para administrar el cannabis, no obstante, su indicación
queda a criterio del médico tratante Para los productos de
dosificación se autoriza la administración por vía oral, tópica, sublingual o
inhalada.
6.3. Combinaciones o mezclas. Los productos medicinales
a base de cannabis no deben contener mezclas o combinaciones con principios
activos, nutrientes y/u otras sustancias con efecto fisiológico o nutricional,
incluyendo compuestos tales como vitaminas, minerales, proteínas, aminoácidos,
plantas, concentrados y extractos de plantas ( distintos a los obtenidos de la
planta de cannabis), ingredientes obtenidos de animales, probióticos,
sustancias bioactivas u otros nutrientes. Lo anterior, no aplica en el caso de
que el ingrediente se utilice como vehículo o excipiente inactivo necesario
para la administración de los cannabinoides.
6.4. Productos medicinales a base de caunabis sujetos a
fiscalización. Productos medicinales a base de cannabis que contengan una
concentración igual o mayor a 0.3 % de THC total en su empaque final para el
caso de productos secos o en su forma de presentación dosificada, tales como
tabletas, cápsulas o similares o por cada mililitro o miligramo en caso de
soluciones o cremas, se considerarán productos sometidos a la fiscalización
establecida en el "Reglamento para el Control de Drogas, Estupefacientes y
Psicotrópicas" vigente.
6.5. Productos medicinales a base de caunabis con CBD. Los
productos formulados con CBD para administrar una dosis mayor a 70 mg/día de
CBD total deben registrarse como productos medicinales a base de cannabis según
lo establecido en el presente reglamento.
7.-
MATERIAS PRIMAS Y MATERIALES. Se establecen las siguientes disposiciones
para las materias primas y materiales relacionados con productos medicinales a
base de cannabis:
7.1. Notificación de material vegetal o derivados
de cannabis al MS. Para la importación, fabricación, distribución y
comercialización del material vegetal o derivados de cáñamo o cannabis
psicoactivo a utilizar para la fabricación de un producto medicinal a base
cannabis se requiere previamente su notificación ante el MS.
7.2. Requisitos para la notificación. Los
requisitos son:
7
.2.1. Comprobante de pago.
7.2.2.
Formulario de solicitud de notificación según el Anexo I de este reglamento.
7.2.3.
En caso de material vegetal o derivados de cannabis importados, documento
apostillado o legalizado donde conste la habilitación del proveedor para la
producción y/o comercialización de material vegetal o ingredientes derivados de
cannabis emitido por la autoridad competente del país de origen.
Cuando
el material vegetal o derivado sea producido a nivel nacional el solicitante
debe indicar en el formulario de notificación, el número de autorización o
licencia de cultivo o de fabricación de derivados según corresponda.
7.2.4.
Especificaciones del material vegetal o derivado de cannabis emitida por el
cultivador o el fabricante que indique nombre científico y parte de la planta utilizada
como materia prima o para la fabricación del derivado, porcentaje de THC y CBD,
material de empaque y condiciones de almacenamiento.
7.2.5.
Descripción del proceso de fabricación del derivado, que incluya un diagrama de
flujo de dicho proceso.
7.2.6.
Certificado o informe de análisis que incluya los ensayos señalados en el
numeral 11.4 del presente reglamento. Dicho certificado debe cumplir con lo
indicado en los numerales 15.1 y 15.2 del presente reglamento según corresponde.
7.3.
Plazo de resolnción del trámite de notificación de material vegetal o
derivado. El MS tendrá un plazo de hasta 15 días naturales para la resolución
de la solicitud de notificación contados a partir de la presentación completa
de la solicitud.
7.4.
Material vegetal o derivado sujetos a fiscalización. El material vegetal o los
derivados de cannabis que posean un porcentaje de THC total igual o superior al
1 % se clasifican como sustancias sujetas a fiscalización establecida en el
"Reglamento para el Control de Drogas Estupefacientes y
Psicotrópicos". Estos materiales deben cumplir con los procedimientos que
determine la Dirección de Drogas y Estupefacientes.
7.5.
Comercialización de material vegetal o derivados. El material vegetal o un
derivado de cannabis que cuente con la notificación correspondiente del MS no
se considera producto terminado, por lo que no debe ser almacenado, distribuido
o comercializado para su venta directa al consumidor. El material vegetal o
derivados de cannabis podrán ser comercializados al consumidor final una vez
que cumpla con la normativa de registro sanitario para productos medicinales a
base de cannabis.
El
material vegetal o un derivado de cannabis que cuente con la notificación
correspondiente del MS solo podrá ser transferido o vendido con el fin de
reformularlo o envasarlo como un producto medicinal a base de cannabis o para
elaborar preparaciones magistrales ( en el caso de derivados), es decir, se
podrá vender o transferir a:
7.5.1.
Otro establecimiento que cuente con autorización o licencia de fabricación para
el tipo de producto en cuestión.
7.5.2.
Para exportación.
7.5.3.
Farmacias que cuenten con autorización o licencia para la fabricación de
preparaciones magistrales a base de cannabis (para el caso de derivados).
7.6. Importación de material vegetal o derivados de cannabis
psicoactivo para uso como materia prima. Podrán importar material
vegetal o derivados de cannabis psicoactivo para su uso como materia prima para
la fabricación de medicamentos y/o productos medicinales a base de cannabis,
solamente las personas fisicas o jurídicas que cuenten con la licencia
correspondiente.
Previo
a la importación y antes de realizar cualquier gestión ante PROCOMER, el
interesado debe tramitar el permiso de importación correspondiente en la
Dirección de Drogas y Estupefacientes según lo establecido en el Decreto
Ejecutivo Nº 44695-MP-S-MAG del 29 de agosto del 2024 "Reglamento a la Ley
Nº 10113, Ley del Cannabis para uso medicinal y terapéutico y del Cáñamo para
uso alimentario e industrial del 02 de marzo del 2022, Reglamento del Cannabis
para Uso Medicinal y Terapéutico".
Una
vez obtenida la autorización por parte de dicha dirección se debe realizar el
trámite ante la VUCE en PROCOMER y presentar los siguientes requisitos:
7.6.1.
FAD indicando en la sección de observaciones lo siguiente:
7.6.1.1.
El número del permiso de importación emitido por la Dirección de Drogas y
Estupefacientes adscrita al Ministerio de Salud.
7.6.1.2.
Número de licencia otorgada.
7.6.1.3.
Número de notificación de la materia prima a importar.
Lo
anterior será verificado por la autoridad de salud ubicada en PROCOMER.
7.6.2.
Copia de la factura y examen previo del agente de aduanas, que debe indicar la
información de los productos a ingresar con el siguiente detalle: nombre de producto,
descripción, cantidad en unidades y presentación comercial.
7.6.3.
Documento de transporte.
7.6.4.
En caso de material vegetal de cannabis psicoactivo, el importador debe cumplir
con los controles fitosanitarios establecidos por el SFE.
Los
documentos indicados en los numerales 7.6.1, 7.6.2 y 7.6.3 deberán ser
presentados para el trámite de desalmacenaje en el sistema VUCE según el
Decreto Ejecutivo Nº 33452-COMEXMAG-H-G-S-MP del 15 de junio del 2006
"Reglamento del Sistema de Ventanilla Única de Comercio Exterior",
asimismo, la autoridad de salud en PROCOMER tendrá un plazo de 10 días
naturales para la resolución del trámite.
7.7.
Importación de material vegetal o derivados de cáñamo para la fabricación de
productos medicinales a base de cannabis. La persona fisica o jurídica que
cuente con la licencia correspondiente para la fabricación de productos
medicinales a base de cannabis o una autorización de importación de material
vegetal y/o derivados de cáñamo para su comercialización como materia prima
para uso alimentario o industrial, deberá realizar el trámite de importación en
la VUCE en PROCOMER y presentar los siguientes requisitos:
7.7.1.
FAD indicando en la sección de observaciones, según corresponda,
7.7.1.1.
Número de licencia u autorización otorgada
7.7.1.2.
Número de notificación de la materia prima a importar.
Lo
anterior será verificado por la autoridad de salud ubicada en PROCOMER.
7.7.2.
Certificado o informe de análisis por cada lote de material vegetal o derivado
de cáñamo a importar en el que se especifique el contenido de THC total. Dicho
certificado debe incluir el nombre y lote del producto importado, nombre del
laboratorio que realiza el análisis, fecha del análisis, firma del profesional
que realiza el análisis, referencia al método de ensayo empleado, el límite de
cuantificación y detección del método utilizado, el contenido de THC debe
reportarse en porcentaje, asimismo, en caso de emitirse en el extranjero debe
cumplir con lo señalado en el numeral 15.2 del presente reglamento.
7.7.3.
Copia de la factura comercial.
7.
7.4. Documento de transporte.
7.7.5.
En caso de material vegetal, el importador debe cumplir con los controles
fitosanitarios establecidos por el SFE.
Los
documentos indicados en los numerales 7.7.1, 7.7.2, 7.7.3 y 7.7.4 deberán ser
presentados para el trámite de desalmacenaje en el sistema VUCE según el
Decreto Ejecutivo Nº 33452-COMEX-MAG-H-G-S-MP del 15 de junio del 2006
"Reglamento del Sistema de Ventanilla Única de Comercio Exterior",
asimismo, la autoridad de salud en PROCOMER tendrá un plazo de 1 O días
naturales para la resolución del trámite.
7.8. Autorización de importación de muestras sin valor comercial
de derivados de caunabis y materiales de referencia. En el caso
de derivados o sustancias que se encuentren en las listas de estupefacientes y
sustancias sicotrópicas sometidas a fiscalización nacional, publicadas en la
página Web oficial del MS, el interesado debe tramitar
previamente el permiso de importación ante la Dirección de Drogas y
Estupefacientes, según lo establecido en el Decreto Ejecutivo Nº 44695-MP-S-MAG
"Reglamento a la Ley Nº 10113, Ley del Cannabis para uso medicinal y
terapéutico y del Cáñamo para uso alimentario e industrial del 02 de marzo del
2022, Reglamento del Cannabis para Uso Medicinal y Terapéutico del 29 de agosto
del 2024".
Se
debe presentar a la Dirección de Drogas y Estupefacientes para el trámite de
autorización de importación lo siguiente, según corresponda:
7.8.1.
Permiso de exportación emitido por la autoridad competente del país de origen.
Lo
anterior será verificado por la autoridad de salud ubicada en PROCOMER.
7
.8.2. Certificado de análisis de la muestra o material por importar que
especifique el contenido de THC total. Dicho certificado en caso de emitirse en
el extranjero debe cumplir con lo señalado en el numeral 15 .2 del presente
reglamento.
7.8.3.
Copia de la factura.
7.8.4.
Documento de transporte y número del PSF del importador.
La
Dirección de Drogas y Estupefacientes tendrá un plazo de 15 días naturales para
la resolución del trámite a partir de la presentación completa de la solicitud.
Cuando
producto de la revisión y verificación de los requisitos solicitados se
comprueba que no se ajusta a lo requerido por el MS, se procederá a emitir por
única vez y por medio de un oficio, la respectiva prevención, en la cual se
indicará al interesado que en el plazo de 10 días hábiles debe completar los
requisitos omitidos en la solicitud o en el trámite, o bien, que en dicho plazo
debe aclarar información necesaria para el estudio y evaluación de esta. Si el
interesado no cumple con lo anterior, se procederá a archivar el expediente y
se notificará al interesado.
Esta
prevención suspende el plazo de resolución que tiene el Ministerio.
Transcurridos los diez días hábiles, o a partir de que el interesado cumpla con
la presentación de los requisitos omitidos según la prevención del Ministerio,
se continuará con el cómputo previsto para resolver.
8.- IMPORTACIÓN, ALMACENAMIENTO Y DISTRIBUCIÓN DE PRODUCTOS
MEDICINALES A BASE DE CANNABIS. Se establecen las siguientes
disposiciones sobre importación, almacenamiento y distribución de productos
medicinales a base de cannabis.
8.1.
Podrán importar, almacenar y distribuir productos medicinales a base de
cannabis con registro sanitario vigente en Costa Rica solamente las personas
fisicas o jurídicas que cuenten con la licencia correspondiente.
8.2.
Las personas fisicas o jurídicas indicadas en el numeral anterior, solo podrán
vender productos medicinales a base de cannabis a farmacias.
8.3 Importación de productos medicinales a base de cannabis. Podrán
importar productos medicinales a base de cannabis, solamente las personas
fisicas o jurídicas que cuenten con la licencia correspondiente. El producto
medicinal a base de cannabis debe contar con registro sanitario vigente en
Costa Rica ante el MS.
En
el caso de productos medicinales a base de cannabis sujetos a fiscalización
previo a la importación y antes de realizar cualquier gestión ante PROCOMER, el
interesado debe tramitar el permiso de importación correspondiente en la
Dirección de Drogas y Estupefacientes según lo establecido en el Decreto
Ejecutivo Nº 44695-MP-S-MAG 29 de agosto del 2024 "Reglamento a la Ley
Nº10113, Ley del Cannabis para uso medicinal y terapéutico y del Cáñamo para
uso alimentario e industrial del 02 de marzo del 2022, Reglamento del Cannabis
para Uso Medicinal y Terapéutico".
Una
vez obtenida la autorización por parte de dicha dirección se debe realizar el
trámite ante la VUCE en PROCOMER y presentar los siguientes requisitos:
8.3.1.
FAD indicando en la sección de observaciones, según corresponda,
8.3.
1.1. Número de licencia otorgada.
8.3.1.2.
Número del permiso de importación emitido por la Dirección de Drogas y
Estupefacientes.
Lo
anterior será verificado por la autoridad de salud ubicada en PROCOMER.
8.3.2.
Lista de empaque de los productos.
8.3.3.
Copia de la factura comercial.
8.3.4.
Documento de transporte.
8.3.5.
Presentar el examen previo aduana!.
Los
documentos indicados en los numerales 8.3.1, 8.3.2, 8.3.3, 8.3.4 y 8.3.5
deberán ser presentados para el trámite de desalmacenaje en el sistema VUCE
según el Decreto Ejecutivo Nº 33452 COMEX-MAG-H-G-S-MP del 15 de junio del 2006
"Reglamento del Sistema de Ventanilla Única de Comercio Exterior",
asimismo, la autoridad de salud en PROCOMER tendrá un plazo de 10 días
naturales para la resolución del trámite.
8.4. Autorización de importación de productos medicinales a base
de cannabis para uso personal. La Dirección de Drogas y
Estupefacientes podrá autorizar la importación de productos medicinales a base
de cannabis para uso personal que no tengan registro sanitario vigente en Costa
Rica, no obstante, en caso de que el producto esté disponible en el comercio no
se autorizará su ingreso.
El
trámite se debe realizar ante la Dirección de Drogas y Estupefacientes y se
deben presentar los siguientes requisitos:
8.4.1.
Copia del certificado médico según formato del Colegio profesional correspondiente,
suscrito por el prescriptor, según el artículo 131 de la Ley General de Salud,
que respalde la necesidad del producto medicinal a base de cannabis a importar
para el paciente en particular.
El
certificado se acepta con fecha de emisión no mayor de tres meses. La cantidad
solicitada debe ser congruente con la prescrita por el profesional y las
necesidades del paciente; no debe sobrepasar la cantidad requerida para 12
meses de tratamiento por cada trámite realizado.
8.4.2.
Receta original según formato del colegio profesional correspondiente, en caso
de que no se incluya la información completa en el certificado, con fecha de
emisión no mayor a tres meses, detallando dosis, forma farmacéutica o
presentación y periodo del tratamiento. La cantidad solicitada debe ser
congruente con la prescrita por el médico y las necesidades del paciente; no
debe sobrepasar la cantidad requerida para 6 meses de tratamiento por cada
trámite realizado.
Para
la autorización a pacientes con seguro médico internacional o control médico en
el extranjero, los requisitos 8.4.1. y 8.4.2 anteriores deben ser emitidos en
el país en que se prescribió el producto medicinal a base de cannabis
originalmente. Cuando estos documentos se encuentren en un idioma diferente al
español se debe aportar la traducción correspondiente.
8.4.3.
En caso de solicitudes de ingreso de cantidades superiores a un mes de
tratamiento de productos medicinales a base de cannabis sujetos a
fiscalización, se requiere tramitar previamente el permiso de importación ante
la Dirección de Drogas y Estupefacientes, según lo establecido en el Decreto
Ejecutivo Nº 44695-MP-S-MAG el 29 de agosto del 2024 "Reglamento a la Ley
Nº 10113, Ley del Cannabis para uso medicinal y terapéutico y del Cáñamo para
uso alimentario e industrial del 02 de marzo del 2022, Reglamento del Cannabis
para Uso Medicinal y Terapéutico".
8.4.4.
La Dirección de Drogas y Estupefaciente podrá autorizar la importación de
productos medicinales a base de cannabis sin registro sanitario vigente en
Costa Rica, en el caso de pacientes con seguro médico internacional o control
médico en el extranjero, para lo cual deberán presentar los mismos requisitos
establecidos en los numerales 8.4.1, 8.4.2 y 8.4.3 del presente reglamento.
La
Dirección de Drogas y Estupefacientes tendrá un plazo de 15 días naturales para
la resolución del trámite a partir de la presentación completa de la solicitud.
Cuando
producto de la revisión y verificación de los requisitos solicitados se
comprueba que no se ajusta a lo requerido por el MS, se procederá a emitir por
única vez y por medio de un oficio, la respectiva prevención, en la cual se
indicará al interesado que en el plazo de 10 días hábiles debe completar los
requisitos omitidos en la solicitud o en el trámite, o bien, que en dicho plazo
debe aclarar información necesaria para el estudio y evaluación de esta. Si el
interesado no cumple con lo anterior, se procederá a archivar el expediente y
se notificará al interesado.
Esta
prevención suspende el plazo de resolución que tiene el Ministerio.
Transcurridos los diez días hábiles, o a partir de que el interesado cumpla con
la presentación de los requisitos omitidos según la prevención del Ministerio,
se continuará con el cómputo previsto para resolver.
9.-
PRESCRIPCIÓN Y DISPENSACIÓN. Se establecen las siguientes disposiciones para la
prescripción
y dispensación de productos medicinales a base de cannabis.
9.1.
Se podrán vender y despachar al público productos medicinales a base de
cannabis debidamente registrados, en farmacias que cuenten con el permiso de
habilitación del MS vigente de acuerdo con lo establecido en el Decreto
Ejecutivo Nº 43432-S del 09 de marzo del 2022 "Reglamento general para
permisos sanitarios de funcionamiento, permisos de habilitación y autorización
para eventos temporales de concentración masiva de personas, otorgados por el
Ministerio de Salud", con regencia al día. Los productos medicinales a
base de cannabis sujetos a fiscalización definidos en el artículo 6.4
del presente reglamento, solo podrán ser prescritos y despachados mediante
recetas digitales en el Sistema Automatizado de Receta Digital cumpliendo con
lo establecido en el Decreto Ejecutivo Nº 39984-S del 01 de setiembre del 2016
"Reglamento de Utilización y Funcionamiento del Sistema Automatizado de
Receta Digital de Psicotrópicos y Estupefacientes. Se prohíbe utilizar
cualquier otro medio para dicho fin, en apego a lo establecido en la Ley
General de Salud, con excepción de los casos de fuerza mayor que se establecen
en el artículo 23 del Decreto Ejecutivo 39984-S del 01 de setiembre del 2016
"Reglamento de Utilización y Funcionamiento del Sistema Automatizado de
Receta Digital de Psicotrópicos y Estupefacientes". Los productos medicinales
a base de cannabis que no son sujetos a fiscalización se deben despachar con
receta médica.
9.2.
Solamente las personas profesionales en medicina humana, en ejercicio legal de
sus profesiones podrán prescribir productos medicinales a base de cannabis.
9.3.
Toda prescripción de productos medicinales a base de cannabis debe responder a
la valoración del paciente, de conformidad con el acto profesional respectivo y
constar debidamente en su expediente clínico.
9.4.
El control y el manejo de productos medicinales a base de cannabis en las
farmacias, así como el despacho de las recetas que se prescriben, corresponderá
personal y exclusivamente a los regentes farmacéuticos.
9.5.
La prescripción de productos medicinales a base de cannabis se permite como
coadyuvante a las terapias estándar, cuando según criterio médico se determine
que los tratamientos convencionales con medicamentos autorizados no están
produciendo los efectos esperados o estén causando efectos adversos relevantes
o si se cumplen las condiciones señaladas en el Anexo II del presente
reglamento.
9.6.
La lista de indicaciones médicas recomendadas para la prescripción de productos
medicinales a base de cannabis es la establecida en el Anexo II del presente
reglamento. El MS actualizará dicha lista en función de la evolución de los
conocimientos técnicos y científicos y se oficializará mediante Decreto
Ejecutivo.
9.7.
La cantidad máxima de productos secos de cannabis que un paciente está
autorizado a poseer en lugar público, para sus propios fines médicos es una
cantidad que es equivalente a la menor de las siguientes:
.
(a) 30 veces la cantidad diaria de productos secos de cannabis indicada en la
receta médica, si tienen más de un documento de registro, 30 veces el total de
las cantidades diarias indicadas en la receta médica, y
.
(b) 150 g de productos secos de cannabis.
10.- REGISTRO SANITARIO. Se establecen las siguientes
disposiciones para el registro sanitario de productos medicinales a base de
cannabis.
10.1. Disposiciones generales para el registro sanitario. Las
disposiciones generales son las siguientes:
10.1.1.
Para la importación, producción, distribución, comercialización y prescripción
de todo producto medicinal a base cannabis se requiere previamente su registro
sanitario ante la UR de la DRPIS del MS.
10.1.2.
El registro sanitario de productos medicinales a base de cannabis tendrá una
vigencia de cinco años, el cual puede ser suspendido o cancelado cuando haya
razones sanitarias de carácter científico, técnico o legal debidamente
justificados.
10.1.3.
Todo documento oficial o legal emitido ante el registro debe legalizarse
cumpliendo con la normativa nacional específica.
10.1.4.
Todo documento oficial o legal requerido para el registro debe estar vigente en
el momento de su presentación. Los documentos oficiales tendrán la validez que
les otorgue la autoridad competente del país donde se emite. En los casos en
los que no se indique la vigencia, esta será de 2 años para los efectos del
trámite de registro a partir de la fecha de emisión.
10.1.5.
Todo documento oficial o legal debe presentarse en original o copia legalizada,
según la legislación nacional vigente. El documento se deberá presentar en
idioma español/castellano o en caso de presentarse en otro idioma, deberá ser
acompañado de su respectiva traducción emitida de conformidad con la
legislación nacional.
10.1.6.
Únicamente si el Certificado de Libre Venta o el Certificado de Buenas
Prácticas de Manufactura están disponibles en bases de datos en la página web
de la autoridad emisora, se permite aportar una certificación notarial de copia
del documento digital.
10.1.7.
En el certificado de libre venta y la documentación presentada para el registro
debe aparecer en forma clara el nombre del fabricante y el nombre del producto.
10.1.8.
Cuando el titular y fabricante del producto son filiales o subsidiarias de una
casa matriz y el certificado de libre venta no indica el nombre del titular del
producto, se debe presentar una carta firmada por el fabricante o casa matriz
donde declare quién es el titular del producto de acuerdo con las definiciones
establecidas en este reglamento, cuando la carta sea emitida en el extranjero
debe estar legalizada o apostillada y con su traducción oficial cuando el
idioma de origen no sea el español, acompañado de su respectiva certificación
notarial firmada digitalmente o según lo establecido en el Decreto Ejecutivo Nº
37988-S del 03 de octubre del 2013 "Reglamento para el Funcionamiento y la
Utilización del Portal "Regístrelo".
10.1.9.
En caso de que la autoridad competente del país de origen o del titular del
producto no emita alguno de los requisitos solicitados en el presente
reglamento, el interesado debe presentar nota debidamente legalizada o
apostillada, dada por dicha autoridad donde haga constar esa situación, o bien
una carta emitida por el titular del producto o su representante legal en la
que se incluya el extracto de la regulación que demuestre claramente que el
requisito correspondiente no se emite y la referencia de dicha normativa para
su verificación. Cuando la nota de la autoridad sea emitida en el extranjero,
debe estar legalizada o apostillada y con su traducción oficial cuando el
idioma de origen no sea el español, acompañada de su respectiva certificación
notarial firmada digitalmente según lo establecido en el Decreto Ejecutivo Nº
37988-S del 03 de octubre del 2013 "Reglamento para el Funcionamiento y la
Utilización del Portal "Regístrelo". En el caso de presentar el
extracto de la regulación, se acepta una carta firmada digitalmente por el titular
del producto o su representante legal que incluya la regulación e indique la
sección específica que demuestra lo solicitado y, cuando aplique, la dirección
web en la cual se pueda corroborar esta información; si la regulación se
encuentra en un idioma diferente al español, la sección específica debe contar
con su traducción oficial según lo establecido en el Decreto Ejecutivo Nº
37988-S del 03 de octubre del 2013 Reglamento para el Funcionamiento y la
Utilización del Portal "Regístrelo".
10.1.10.
No se permiten correcciones en las certificaciones o en los documentos
oficiales presentados, a menos que estén sustentadas por la misma instancia que
emitió el documento original.
10.1.11.
En aquellos casos en que aplique y para efectos del registro de un producto
medicinal a base cannabis específico, se permitirá que el solicitante haga
referencia a documentos originales vigentes que consten en archivos del MS. En
este caso el solicitante debe hacer referencia de la gestión en la cual se
entregó el documento original, presentando fotocopia simple del mismo.
10.1.12.
Comprenden un mismo registro sanitario las diferentes presentaciones
comerciales de productos medicinales a base de cannabis con la misma
concentración de CBD y/o THC y la misma forma farmacéutica.
10.2. Requisitos de registro sanitario. Los
requisitos son:
10.2.1.
Comprobante de pago del trámite de registro.
10.2.2.
Solicitud de registro sanitario firmada por el profesional responsable, conteniendo
la información detallada Anexo III del presente reglamento.
10.2.3.
Poderes que acrediten la representación legal y/o técnica otorgada por el
titular a la (s) persona (s) natural (es) o jurídica (s) (original o fotocopia
autenticada del documento).
10.2.4.
Certificado de libre venta del producto, emitido por la autoridad competente
del país de origen o procedencia.
10.2.5.
En caso de productos secos de cannabis importados, se debe presentar un
certificado emitido por la autoridad competente de que el producto proviene de
cultivos lícitos en el país de origen.
10.2.6.
Certificado de Buenas Prácticas de Manufactura de cada uno de los
establecimientos que intervienen en la fabricación del producto, para la forma
farmacéutica y tipo de producto a registrar, extendido por la Autoridad
Competente del país o países en donde se lleva a cabo el proceso de
fabricación, o en su lugar se deberá presentar un documento equivalente al
certificado de Buenas Prácticas de Manufactura, extendido por la Autoridad Competente
o por la Autoridad Reguladora en el que se indique que realiza inspecciones
periódicas al establecimiento, pero que no se extiende el certificado de Buenas
Prácticas de Manufactura.
10.2.7.
Contrato de fabricación a terceros o en su defecto el extracto relativo de las
partes del contrato de fabricación, cuando aplique, en original o fotocopia
autenticada o certificada del documento legalizado, que contenga al menos la
siguiente información:
10.2.7.1.
Firmado por el titular y el fabricante en forma conjunta o separado.
10.2.7.2.
Compromiso de cumplimiento de buenas prácticas de manufactura.
10.2.7.3.
Establecer las condiciones de producción, análisis, cuando aplique o cualquier
otra gestión técnica relacionada con estos.
10.2.7.4.
Debe describir el manejo de materias primas, material de acondicionamiento,
producto a granel y producto terminado, en caso de que estos materiales o
productos sean rechazados.
10.2.7.5.
permitir el ingreso del contratante a las instalaciones del contratista
(contratado) para auditorías.
10.2.7.6.
Listar los productos o servicios objeto del contrato.
10.2.8.
Composición del producto en caso de productos secos o fórmula cualitativa y
cuantitativa completa del producto por unidad de dosis para productos
dosificados. Se debe presentar en original firmada por el profesional
responsable del laboratorio fabricante, indicando según corresponda Jo
siguiente:
10.2.8.1.
Nombre y concentración de los cannabinoides que contiene (CBD y/o THC). Se debe
indicar si el porcentaje se ha calculado en peso o volumen.
10.2.8.2.
Disolvente utilizado, en extractos líquidos. Si el solvente es etanol debe
declarar el porcentaje.
10.2.8.3.
Relación droga/disolvente o excipiente, en caso de extracto o la
estandarización declarada por el fabricante del extracto.
10.2.8.4.
Todos los excipientes del producto ya sea seco (si aplica) o dosificado deben
ser descritos con su denominación internacionalmente aceptada.
10.2.8.5.
Las unidades de cada componente deben estar dadas según el sistema
internacional de medidas (SI).
10.2.8.6.
Composición cualitativa de las cápsulas vacías. Declarar el número de cápsula
utilizada.
10.2.8.7.
Composición de las tintas de impresión en las cápsulas, grageas y tabletas
recubiertas.
10.2.8.8.
Declaración cualitativa de los disolventes orgánicos clase 2 o 3, utilizados en
el proceso de fabricación.
10.2.9.
Ficha técnica del producto terminado.
La
ficha técnica del producto medicinal a base de cannabis a registrar debe
contener la siguiente información:
10.2.9.1.
Nombre del producto.
10.2.9
.2. Composición:
10.2.9.2.1.
Nombre científico de la planta indicando el órgano utilizado.
10.2.9.2.2.
Cantidad de CBD y/o TI-IC en el producto o por unidad de dosis para productos
de dosificación.
10.2.9.3.
Descripción del proceso de fabricación del producto, que incluya un diagrama de
flujo de dicho proceso.
10.2.9.4.
Forma farmacéutica, si corresponde,
10.2.9.5.
Contraindicaciones
10.2.9.6.
Precauciones y advertencias.
10.2.9.7.
Interacciones.
10.2.9.8.
Efectos adversos.
10.2.9.9.
Modo de empleo y vía de administración.
10.2.9
.10. Descripción del material del empaque primario utilizado (por ejemplo,
polímeros, tipos de vidrio), contenedores, sellos, cierres y cualquier
dispositivo de entrega suministrado con el producto.
10.2.9.11.
Recomendación en caso de sobredosis o abuso, cuando aplique.
10.2.9.12.
Referencias bibliográficas.
10.2.9.13.
Fecha de revisión de la ficha técnica.
10.2.10.
Metodología analítica, en caso de no ser un método normalizado debe presentar
el informe de validación del método de ensayo.
10.2.11.
Especificaciones del producto terminado, las cuales deben coincidir con las
indicadas en el numeral 11.4 del presente reglamento.
10.2.12.
Etiquetado de envase o empaque primario, secundario e inserto.
10.2.13.
Informe de estudio de estabilidad.
Nota 1: La fecha
de vencimiento a indicar en un producto medicinal a base de cannabis será de
doce meses a partir del día de su envasado, excepto si existen datos que
indiquen que el producto es estable por otro período de tiempo. Para lo
anterior, se debe presentar informe de estudio de estabilidad conforme a lo
establecido en el Decreto Ejecutivo Nº 36638-COMEX-S-MEIC del 30 de marzo del
2011 "Publicación de la Resolución No. 2562010 (COMIECO-LIX) de fecha 13 de
diciembre del 2010: Reglamento Técnico Centroamericano RTCA 11.01.04: 10
Productos Farmacéuticos. Estudios de Estabilidad de Medicamentos para Uso
Humano", los estudios de estabilidad deben incluir como mínimo las
siguientes pruebas, las cuales deben cumplir con las especificaciones
establecidas en el numeral 11.4 del presente reglamento.
.
Contaminación microbiológica
.
Pérdida por secado ( cuando corresponda)
.
Ensayo de cannabinoides.
10.3. Requisitos para la renovación del registro sanitario. La
renovación del registro sanitario de un producto medicinal a base de cannabis
deberá gestionarse al menos tres meses antes de su vencimiento.
Una
vez vencido el registro sanitario no se podrá comercializar el producto
debiendo tramitarse como registro nuevo.
Si
durante los 6 meses posteriores al vencimiento del registro del producto, el
interesado solicita se le mantenga el número asignado presentando la causa
justificada, el MS le mantendrá el número original, sin embargo, durante este
período, no podrá comercializarlo.
No
se otorgará la renovación, hasta haber aprobado los cambios post registro
solicitados.
10.3.1. Cuando el producto mantiene la información y
características que han sido aprobadas durante la vigencia del registro, al
solicitar la renovación deben presentar:
10.3.1.1
Comprobante de pago de la renovación de registro.
10.3.1.2.
Solicitud de renovación de registro sanitario firmada y sellada por el
profesional responsable conteniendo la información detallada en el Anexo III.
10.3.1.3.
Declaración jurada emitida por el titular o su representante legal o por el
profesional responsable del registro mediante poder emitido por el titular del
producto, que indique que la información y características del producto no han
variado desde la última solicitud de modificación presentada ante el MS.
10.3.1.4.
Certificado de libre venta del producto, emitido por la autoridad competente el
país de origen o procedencia.
10.3.1.5.
En caso de productos secos de cannabis importados, además se debe presentar un
certificado emitido por la autoridad competente de que el producto proviene de
cultivos lícitos en el país de origen.
10.3.1.6.
Certificado de Buenas Prácticas de Manufactura de cada uno de los establecimientos
que intervienen en la fabricación del producto, para la forma farmacéutica y
tipo de producto a registrar, extendido por la Autoridad Competente del país o
países en donde se lleva a cabo el proceso de fabricación, o en su lugar se
deberá presentar un documento equivalente al certificado de Buenas Prácticas de
Manufactura, extendido por la Autoridad Competente o por la Autoridad
Reguladora en el que se indique que realiza inspecciones periódicas al
establecimiento, pero que no se extiende el certificado de Buenas Prácticas de
Manufactura.
10.3.1.7
Informe del estudio de estabilidad que confirme el período de vida útil
aprobado. Ver Nota 1 indicada en el numeral en el numeral 10.2.13.
10.3.2. En los casos que el titular de un producto
solicite modificaciones en el registro sanitario en forma simultánea a la
renovación:
Debe
cumplir los siguientes requisitos:
10.3.2.1.
Comprobante de pago.
10.3.2.2.
Solicitud de renovación de registro sanitario y de las modificaciones firmada y
por el profesional responsable, conteniendo la información detallada Anexo II.
10.3
.2.3. Poderes que acrediten la representación legal y/o técnica otorgada por el
titular a la (s) persona (s) natural (es) o jurídica (s) (original o fotocopia
autenticada del documento).
10.3.2.4
Certificado de libre venta del producto, emitido por la autoridad competente
del país de origen o procedencia.
10.3.2.5.
En caso de productos secos de cannabis importados, además se debe presentar un
certificado emitido por la autoridad competente de que el producto proviene de
cultivos lícitos en el país de origen.
10.3.2.6.
Certificado de buenas prácticas manufactura, conforme a lo establecido en el
numeral 10.2.6 de requisitos de registro.
10.3.2.7.
Contrato de fabricación, cuando aplique, de acuerdo con el numeral 10.2.7 de
requisitos de registro.
10.3.2.8.
Composición del producto en caso de productos secos o fórmula cualitativa y
cuantitativa completa del producto por unidad de dosis para productos
dosificados, conforme el numeral 10.2.8 requisitos de registro.
10.3.2.9.
Especificaciones del producto terminado.
10.3.2.10.
Etiquetado de envase o empaque primario, secundario e inserto. Cuando el
producto no ha sido comercializado, se aceptará el proyecto del arte de
impresión del empaque primario y secundario en idioma español, acompañado de
una declaración jurada del titular del producto que indique que el producto no
ha sido comercializado.
10.3.2.11.
Informe de Estudio de Estabilidad que confirme el periodo de vida útil. Ver
Nota 1 indicada en el numeral en el numeral 10.2.13.
10.3.2.12.
Según la modificación solicitada deberá presentar los documentos según Anexo IV
del presente reglamento.
10.4. Causas de no otorgamiento del registro sanitario. No se
emitirán el registro sanitario de un producto cuando:
10.4.1.
No cumpla con los requisitos establecidos.
10.4.2.
La fórmula contenga ingredientes reportados como no seguros o en cantidades y
vías no permitidas.
10.4.3.
La fórmula o composición de producto contenga sustancias señalas en el artículo
6.3 del presente reglamento.
10.5. Causas de cancelación del registro sanitario. Se
cancelará el registro sanitario de un producto cuando:
10.5
.1. Se compruebe que el producto resulte ser nocivo o no seguro en las
condiciones normales de uso.
10.5.2.
Por falsificación o alteración de los documentos presentados ante el MS.
10.5.3.
Cuando se demuestre que el producto no tiene la composición cuantitativa o
cualitativa autorizada o cuando se incumplan las garantías de calidad y
estabilidad declaradas en el expediente, siguiendo el debido proceso.
10.5.4.
Cuando lo solicite el titular del producto.
10.6. Modificaciones al registro sanitario. Toda
modificación en la información posterior al registro sanitario se ajustará a lo
establecido en el Anexo IV de este reglamento.
Cuando
existan cambios en la forma farmacéutica y/o concentración de CBD y/o THC se
debe tramitar un nuevo registro.
10.7. Plazos de resolución de trámites de registro sanitario. Se
establecen los siguientes plazos de resolución para trámites de registro
sanitario:
10.7.1. Registro sanitario por primera vez. Para el
registro de productos medicinales a base de cannabis el MS dispondrá de un
plazo de hasta treinta (30) días naturales a partir de la presentación completa
de la solicitud correspondiente, para aprobarla y emitir el registro sanitario
o bien para rechazarla.
10.7.2. Renovaciones del registro sanitario sin cambios
(declaración jurada). Para las solicitudes de renovaciones del
registro sanitario de productos medicinales a base de cannabis con declaración
jurada de que no hay cambios en el producto, el MS dispondrá de un plazo de hasta
veintidós (22) días naturales a partir de la presentación completa de la
solicitud correspondiente, para aprobarla y emitir el registro sanitario o bien
para rechazarla.
10.7.3. Renovaciones del registro sanitario con cambios. Para las
solicitudes de renovación del registro sanitario de productos medicinales a
base de cannabis con cambios en el producto, el MS dispondrá de un plazo de
hasta treinta (30) días naturales a partir de la presentación completa de la
solicitud correspondiente, para aprobarla y emitir el registro sanitario o bien
para rechazarla.
10.7.4. Modificaciones o cambios posteriores al registro
sanitario. Para las solicitudes de modificaciones o cambios post registro
sanitario el MS dispondrá de un plazo de hasta treinta (30) días naturales a
partir de la presentación completa de la solicitud correspondiente, para
aprobarla y emitir el registro sanitario o bien para rechazarla.
11.- ESPECIFICACIONES. Se establecen las siguientes
disposiciones relacionadas con especificaciones de productos medicinales a base
de cannabis.
11.1. Requisitos para la fabricación e importación de productos
medicinales a base de cannabis. Un establecimiento dedicado a la
fabricación o importación de productos medicinales a base de cannabis debe
contar un permiso sanitario vigente para la actividad de acuerdo con lo
establecido en el Decreto Ejecutivo Nº 43432-S del 09 de marzo del 2022 "Reglamento
general para permisos sanitarios de funcionamiento, permisos de habilitación y
autorización para eventos temporales de concentración masiva de personas,
otorgados por el Ministerio de Salud" y la autorización o licencia
correspondiente según lo establecido en el Decreto Ejecutivo Nº 44585-MP-MAG-S
del 12 de julio del 2024 Reglamento a la Ley Nº 10113, "Ley del Cannabis
para uso medicinal y terapéutico y del Cáñamo para uso alimentario e industrial
del 02 de marzo del 2022" , Reglamento del Cáñamo para Uso Alimentario e
Industrial o Uso Medicinal o Terapéutico y el Decreto Ejecutivo No.
44695-MP-S-MAG del 29 de agosto del 2024 Reglamento a la Ley Nº 10113 del 02 de
marzo del 2022 " Ley del Cannabis para uso medicinal y terapéutico y del
Cáñamo para uso alimentario e industrial" Reglamento del Cannabis para Uso
Medicinal y Terapéutico.
Los
establecimientos que fabriquen productos medicinales a base de cannabis deben
cumplir con las BPM vigentes para productos naturales medicinales Decreto
Ejecutivo Nº 42918-COMEX-MEIC-S del 18 de enero del
2021 "Publicación de la resolución Nº 423-2020(COMIECO-XC) fecha de 30 de
abril del 2020 y sus Anexos: "Reglamento Técnico Centroamericano RTCA
11.03.69:13 Productos Farmacéuticos. Productos Naturales Medicinales para uso humano.
Buenas Prácticas de Manufactura y su guía de verificación".
11.2. Especificaciones de calidad del material vegetal utilizado
como materia prima. Cada lote de una cosecha de material vegetal que reciba un
establecimiento con autorización o licencia para la fabricación de derivados o
productos medicinales de cannabis ya sea cultivado en el país o importado, debe
cumplir con las especificaciones y requisitos en materia fitosanitaria, de
acuerdo con la normativa vigente aplicable y las disposiciones que establezca
el MAG o sus instituciones adscritas. Para cada lote recibido el
establecimiento debe contar con la documentación que demuestre que el material
vegetal cumple con las especificaciones establecidas.
11.3. Registro de ingreso de material vegetal. Cada lote
de material vegetal cultivado a nivel nacional o importado que reciba un
establecimiento con autorización o licencia para la fabricación de derivados o
productos medicinales a base de cannabis debe contar con un registro de ingreso
en donde se consigne la siguiente información.
11.3.1.
Nombre, dirección del cultivador o proveedor (en caso de ser importado) y
número de licencia de cultivo otorgado por el MAG según corresponda.
11.3.2.
Fecha en que se recibió la cosecha.
11.3.3.
Lote y sección o bloque de cultivo, origen, variedad utilizada, origen de la
semilla.
11.3.4.
Fecha de cosecha.
11.3.5.
Número de identificación interno del material recibido.
11.3.6.
Cantidad de material recibido.
11.3.7.
Identificación científica del material vegetal recibido. Género, especie,
subespecie, variedad u otros).
11.3.8.
Parte (s) de la planta.
11.3.9.
Datos generales del material vegetal: sustancias químicas de uso agrícola
aplicadas durante el desarrollo del cultivo, descripción del tratamiento post
cosecha y secado, forma de encasado, condiciones de almacenamiento y transporte.
11.3.10.
Número i referencia a los certificados de análisis.
11.4.
Ensayos y especificaciones de calidad de derivados de cannabis y productos
medicinales a base de cannabis. Cada lote de derivados de cannabis y
productos medicinales a base de cannabis debe cumplir con las siguientes
ensayos y especificaciones:
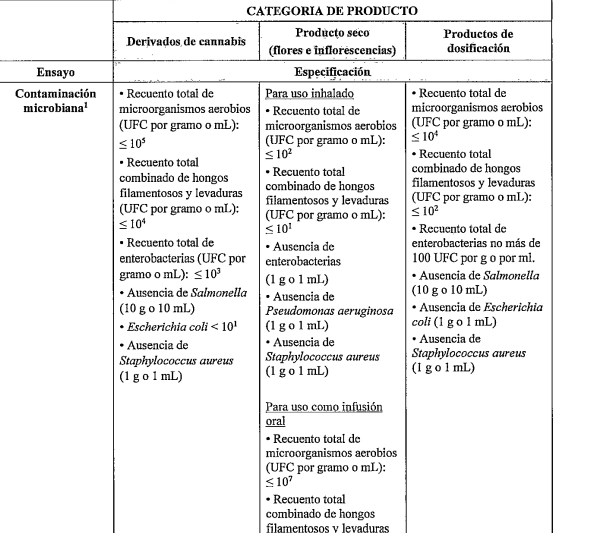
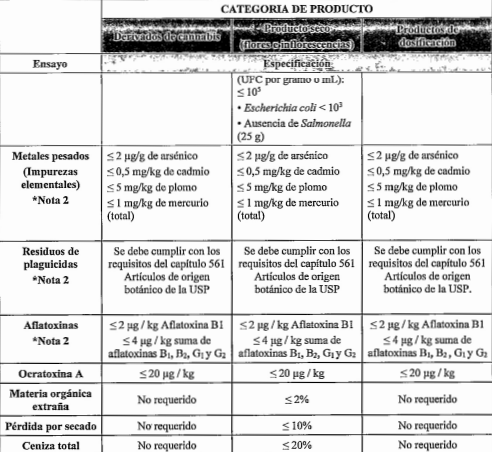

1 Los
criterios de aceptación, en relación con la calidad microbiológica, se
interpreta como:
(a) 10 1
UFC: el recuento máximo aceptable es 20;
(b) 10 2
UFC: el recuento máximo aceptable es 200;
(c) 10 2
UFC: el recuento máximo aceptable es 2000, y así sucesivamente.
Nota 2: Para
productos dosificados el fabricante nacional podrá demostrar cumplimiento si se
realiza el ensayo en el derivado utilizado en la fabricación del lote
correspondiente.
Nota 3: Los
solventes residuales se analizan si se usan técnicas de extracción o basadas en
solventes o si emplean solventes durante la fabricación.
12.-MARCADO Y ETIQUETADO. Requisitos de etiquetado
de productos medicinales a base de cannabis.
12.1. Condiciones generales del etiquetado de productos
medicinales a base de cannabis.
La
información de la etiqueta o rótulo en condiciones normales de manipulación del
producto debe mantenerse fácilmente legible, estar redactada en idioma
castellano/español. El uso simultáneo de otros idiomas será aceptado siempre y
cuando la información sea la misma.
Las
etiquetas podrán ser de papel o de cualquier otro material que pueda ser
adherido a los envases o empaques o bien de impresión permanente sobre los mismos;
siempre y cuando este proceso de impresión no altere la fotegridad del envase o
empaque sobre el cual se realiza dicha impresión.
La
impresión de las etiquetas que se adhieran al envase o empaque podrá estar en
el reverso de estas, siempre que sea claramente visible y legible a través del
envase o empaque con su contenido.
Para
efectos de etiquetado las burbujas, cunas, bandejas y otros aditamentos, no se
consideran envase o empaque secundario.
Si
el producto se va a comercializar sin el envase o empaque secundario, el
etiquetado del envase o empaque primario debe cumplir con todos los requisitos
indicados para el envase o empaque secundario.
12.2. Etiquetado de productos medicinales a base de cannabis. El
etiquetado de los productos medicinales a base de cannabis debe cumplir lo
siguiente:
12.2.1 Etiquetado del envase/ empaque primario. La
información que deberá llevar la etiqueta del envase o empaque primario del
producto, cuando no tenga empaque o envase secundario, es la siguiente:
12.2.1.1.
Nombre del producto.
12.2.1.2.
Forma farmacéutica.
12.2.1.3.
Modo de empleo.
12.2.1.4.
Concentración de CBD y/o THC, por forma dosificada cuando corresponda.
12.2.1.5.
Número de registro sanitario.
12.2.1.6.
Nombre del laboratorio fabricante y país de origen. En caso de fabricación por
terceros, se debe incluir nombre y país de origen de los laboratorios
involucrados en los diferentes procesos de fabricación.
12.2.1.7.
Cantidad o volumen neto del producto terminado en el envase declarado en el
Sistema Internacional de Unidades.
12.2.1.8.
Número de lote.
12.2.1.9.
Condiciones de almacenamiento.
12.2.1.10.
Fecha de vencimiento.
12.2.1.11.
Contraindicaciones y advertencias si proceden.
12.2.1.12.
Leyendas generales.
12.2.1.13.
Leyendas especiales, si proceden.
12.2.1.14.
Vía de administración
En
caso de que el producto se dispense al usuario con su empaque o envase
secundario o con inserto, la información indispensable que debe incluir en el
envase o empaque primario debe ser:
12.2.1.15.
Nombre del producto.
12.2.1.16.
Número de lote.
12.2.1.17.
Fecha de vencimiento.
12.2.1.18.
Nombre o logotipo del laboratorio fabricante.
12.2.2. Etiquetado del envase/ empaque secnndario de productos
medicinales a base de cannabis. La información que debe llevar la
etiqueta del envase o empaque secundario del producto es la siguiente:
12.2.2.1.
Nombre del producto.
12.2.2.2.
Forma farmacéutica
12.2.2.3.
Modo de empleo.
12.2.2.4.
Concentración de CBD y/o THC, por forma dosificada cuando corresponda.
12.2.2.5.
Número de registro sanitario.
12.2.2.6.
Nombre del laboratorio fabricante y país de origen. En caso de fabricación por
terceros, se debe incluir nombre y país de origen de los laboratorios
involucrados en los diferentes procesos de fabricación.
12.2.2.7.
Cantidad o volumen neto del producto terminado en el envase declarado en el
Sistema Internacional de Unidades.
12.2.2.8.
Número de lote.
12.2.2.9.
Condiciones de almacenamiento.
12.2.2.10.
Fecha de vencimiento.
12.2.2.11.
Contraindicaciones y advertencias (si proceden).
12.2.2.12.
Interacciones (si proceden).
12.2.2.13.
Efectos adversos (si proceden).
12.2.2.14.
Leyendas generales.
12.2.2.15.
Leyendas especiales, si proceden.
12.2.2.16.
Vía de administración.
Si
la totalidad de la información exigida en los numerales 12.2.l y 12.2.2 no
puede ser consignada en la etiqueta o empaque, debe incluirse utilizando
inserto, instructivo o prospecto.
12.3. Leyendas generales en productos medicinales a base de
cannabis. Las leyendas que deben figurar en el etiquetado del producto
medicinal a base de cannabis se citan a continuación.
12.3.1.
Leyendas generales:
12.3.1.1.
"Manténgase fuera del alcance de los niños".
12.3.1.2.
"El uso de este producto durante el embarazo o en periodo de lactancia
puede ser perjudicial para la salud".
12.3.1.3.
"No se ha aprobado la seguridad y eficacia de este producto. Consulte a su
médico antes de usar este producto con suplementos a la dieta o
medicamentos".
12.3.1.4.
"Venta Bajo Receta Médica".
12.3.1.5.
"Precaución, puede crear dependencia" o una frase similar
12.4. Etiquetado de material vegetal. Cada lote
de material vegetal que reciba un fabricante de derivados de cannabis o de
productos medicinales a base de cannabis de parte de un establecimiento
nacional que cuente con autorización o licencia de cultivo o importado, debe
tener una etiqueta con la siguiente información en idioma español:
12.4.1.
Tipo de producto: cáñamo o cannabis psicoactivo.
12.4.2.
Número de identificación interno del lote recibido.
12.4.3.
Número de sección o bloque donde se cosechó.
12.4.4.
Nombre del cultivador o proveedor (si se importa).
12.4.5.
País de origen.
12.4.6.
Condiciones de almacenamiento.
12.4.7.
Porcentaje de CBD total y/o THC total.
En
caso de material vegetal importado con etiquetado en un idioma diferente al
español, se permite colocar una etiqueta complementaria para incluir la
información señalada en idioma español.
12.5. Etiquetado de derivados. El
etiquetado de los derivados de cannabis utilizados para la fabricación de
productos medicinales a base de cannabis importados o de fabricación nacional,
debe contener la siguiente información en idioma español:
12.5
.1. Nombre y/o marca comercial del derivado.
12.5.2.
Identificación del lote.
12.5.3.
Tipo de derivado (cáñamo o cannabis psicoactivo).
12.5.4.
Contenido neto.
12.5.5.
Razón social y dirección completa del fabricante o proveedor (en caso de ser
importado), según corresponda.
12.5.6.
Fórmula cualitativa.
12.5.7.
Uso.
12.5.8.
País de fabricación del derivado.
12.5.9.
Condiciones de almacenamiento.
12.5.10.
Porcentaje de THC total y CBD total.
12.5.11.
Fecha de vencimiento.
En
caso de derivados importados con etiquetado en un idioma diferente al español,
se permite colocar una etiqueta complementaria para incluir la información
señalada en idioma español.
13.- TOMA DE MUESTRA Y MUESTREO. Se
establecen las siguientes disposiciones para el muestreo de productos
medicinales a base de cannabis.
13.1. Representatividad y custodia de la muestra. La
muestra por analizar de productos medicinales a base de cannabis debe ser
representativa del lote fabricado, por lo que cada fabricante
o importador debe elaborar e implementar procedimientos de muestreo basados en
fundamentos científicos, como tablas militares o monografias de muestreo de las
farmacopeas oficiales vigentes establecidas en Decreto Ejecutivo Nº
43259-COMEX-S-MEIC del 27 de setiembre del 2021 Publicación de la Resolución Nº
446-2021 (COMIECO-XCIV) de fecha 28 de abril de 2021 y sus Anexos: "Anexo
I: Reglamento Técnico Centroamericano RTCA 11.03.59:18 Productos Farmacéuticos.
Medicamentos para Uso Humano. Requisitos de Registro Sanitario" y
"Anexo II: Reconocimiento Mutuo de Registro Sanitario de Medicamentos para
Uso Humano"
Dicho
procedimiento debe describir lo siguiente: método de muestreo, equipo, cantidad
de muestra, recipiente de muestreo, identificación del contenido, condiciones
de almacenamiento e instrucciones de limpieza y almacenamiento. Cualquier
cambio al procedimiento se documentará para inspección en cualquier momento.
Las
muestras de derivados de cannabis y productos medicinales a base de cannabis
enviadas a un laboratorio con actividades acreditadas en el análisis o ensayo
específico, deben ir acompañadas de un formulario de cadena de custodia, que
debe incluir el número y tipos de contenedores, cantidad de muestra, método o
procedimiento de muestreo, la identidad del muestreador o responsable del
muestreo, fecha de recolección de la muestra, las condiciones de almacenamiento
o conservación, la identidad de los transportistas y análisis solicitados.
14. MÉTODOS DE ANÁLISIS. Se establecen las siguientes
disposiciones relacionadas con los métodos de análisis de productos medicinales
a base de cannabis.
14.1. Métodos de ensayo. Los laboratorios de ensayo deben
garantizar que el método utilizado en la verificación de los requisitos
especificados en el presente reglamento cumpla con los requisitos particulares
para el uso específico y que sean métodos de ensayo basados en métodos
normalizados (F ARMACOPEICOS, INTECO, ISO, ASTM, AOAC) verificados o en su
defecto en métodos de ensayo validados no normalizados o de organismos
competentes en este campo.
El
método de ensayo para determinar el contenido de cannabinoides (se aceptan
espectroscopia de masas en combinación con cromatografia líquida,
espectroscopia de masas en combinación con cromatografia de gases y
cromatografia líquida) debe tener la capacidad de separar los diferentes carmabinoides,
en particular: THC, THC-A, CBD y CBD-A, esto es, debe diferenciar entre el
cannabinoide y su forma ácida. Los resultados de contenido o concentración de
los diferentes cannabinoides en cada producto deben reflejar el contenido de
THC total y CBD total en base seca según corresponda, es decir, la suma del
contenido del cannabinoide y su ácido correspondiente (THCA y CBDA
respectivamente). Para el THC, el contenido total se refiere a la suma de
/',9-THC y ácido /',9-THCA, expresados como /',9 -THC. Para el CBD, el
contenido total se refiere a la suma de CBD y ácido CBDA, expresados como CBD.
Las diferentes formas ácidas se deben calcular aplicando el factor de
conversión correspondiente.
14.2. Materiales de referencia. Las
características químicas del producto deben compararse contra materiales de
referencia certificados. Se deben utilizar aquellos establecidos en el método
de ensayo seleccionado.
15.- PROCEDIMIENTO DE EVALUACIÓN DE LA CONFORMIDAD. Se
establecen las siguientes disposiciones relacionadas con el procedimiento de
evaluación de la conformidad de los productos medicinales a base de cannabis.
15.1 Acreditación de laboratorios de ensayo. Las
pruebas de calidad a realizar para cumplir con lo establecido en los numerales
15.3. y 15.4 del presente reglamento, deben ser realizadas por un laboratorio
de ensayo con el alcance específico del ensayo que corresponda, debidamente
acreditado en la norma INTE-ISO/IEC 17025 "Requisitos generales para la
competencia de los laboratorios de ensayo y de calibración" en su versión
vigente, dicha acreditación debe ser emitida o reconocida por el ECA conforme a
lo establecido en la Ley Nº 10473 del 24 de abril de 2024 "Ley del Sistema
Nacional para la Calidad". En el caso de reconocimiento el interesado debe
seguir el procedimiento correspondiente establecido por el ECA y publicado su
página Web (www.eca.or.cr).
En
caso de que se verifique de acuerdo con la información publicada en la página
Web del ECA, que no existen laboratorios de ensayo acreditados con el alcance
específico del ensayo que corresponda, se aceptarán certificados de análisis de
laboratorios de ensayo que tengan al menos un ensayo acreditado bajo la técnica
analítica que se utilice para la determinación que corresponda.
15.2. Informes de análisis emitidos en el extranjero. En caso
de que un certificado o informe de análisis sea emitido en el extranjero, el
mismo debe estar legalizado o apostillado y con traducción oficial en caso de
que se encuentre en idioma distinto al español.
15.3. Controles de productos comercializados. Para
demostrar la conformidad de los productos medicinales a base de cannabis con
las disposiciones del presente reglamento, el fabricante o importador, debe
elaborar y mantener en sus archivos por cada lote a comercializador, un informe
de análisis firmado por el responsable de la liberación del lote, en el que se
especificará lo siguiente:
15.3.1.
El nombre del producto de cannabis (nombre "genérico") y su
composición
15.3.2.
Fecha de fabricación y de vencimiento del lote.
15.3.3.
Número de lote.
15.3.4.
Tamaño y el número de unidades del lote.
15.3.5.
Nombre del laboratorio fabricante.
15.3.6.
Las concentraciones de CBD total y/o THC total según corresponda.
15.3.7.
Especificaciones y resultados de pruebas según las especificaciones
establecidas.
15.3.8.
Nombre del laboratorio que realiza los análisis y referencia al informe de
análisis respectivo.
15.3.9.
Condiciones de almacenamiento.
15.3.10.
El número o referencia del formulario de cadena de custodia señalado en el
numeral 13.1 del presente reglamento.
15.4. Declaración de la conformidad de primera parte. Se
establecen las siguientes disposiciones para la declaración de la conformidad
de primera parte de los productos medicinales a base de cannabis:
15.4.1.
El titular del registro del producto medicinal a base de cannabis previo a
comercializar el producto debe presentar al MS una declaración de primera parte
que garantice el cumplimiento del reglamento técnico conforme el Anexo V del
presente reglamento. Se deberá presentar nuevamente la declaración indicada al
comercializar el primer lote una vez aprobado e implementados los siguientes
cambios post registro:
15.4.1.1
Cambio de fabricante y de país de origen.
15.4.1.2.
Cambio de excipientes o cambio en la concentración de estos.
15.4.2.
Dicha declaración debe estar respaldada en los resultados del ensayo de un lote
del producto medicinal a base de cannabis, emitido por un laboratorio de ensayo
acreditado o reconocido por ECA, designado por el titular del registro
sanitario.
15.4.3.
Los resultados de los análisis del lote deben demostrar el cumplimiento de los
requisitos establecidos en el presente reglamento, dichos informes de análisis
deben cumplir con lo indicado en los numerales 15.1 o 15.2 del presente
reglamento según corresponda. El informe emitido por el laboratorio de ensayo
debe incluir la siguiente información:
15.4.3.1.
Un título, que indique "Informe de ensayo".
15.4.3.2.
El nombre y la dirección del laboratorio y el lugar en donde se realizaron los
ensayos.
15.4.3.3.
Una identificación única y clara del informe en donde se le asigne un número de
serie o codificación unívoca;
15.4.3.4.
El nombre y la dirección del cliente. Debe establecerse el nombre del cliente
(fisico o jurídico) que solicita el ensayo y su dirección exacta (si el
análisis se contrata a un tercero).
15.4.3.5.
La identificación o referencia del método de ensayo utilizado;
15.4.3.6.
Una descripción inequívoca del producto medicinal a base de cannabis muestreado
o analizado en donde se incluya el nombre del fabricante o importador, el
número de serie o lote, fecha de vencimiento y la fecha de vencimiento, según
aplique;
15.4.3.7.
La fecha de recepción de las muestras sometidas a ensayo;
15.4.3.8.
La fecha de ejecución del ensayo;
15.4.3.9.
El número o referencia del formulario de cadena de custodia señalado en el
numeral 13.1 del presente reglamento.
15.4.3.10.
Los resultados de los ensayos con sus unidades de medida, según lo dispuesto en
el presente reglamento; el contenido del cannabinoide debe reportarse en
porcentaje peso/peso, peso/volumen y/o en unidades del sistema internacional.
15.4.3
.11. El número de réplicas o repeticiones para cada determinación de ensayo;
15.4.3.12.
Una declaración sobre la incertidumbre expandida de la medición. La cual deberá
indicar el factor de cobertura con un nivel estadístico de confianza y el
método empleado para estimar dicha incertidumbre; para el caso de la
determinación de un cannabinoide se debe indicar el límite de cuantificación
y/o detección.
15.4.3.13.
El nombre, función y firma de la persona o personas que autorizan el informe de
ensayo;
15.4.3.14.
El número de página para cada una de las páginas del informe de ensayo y el
número total de páginas de las cuales está constituido;
15.4.3.15.
Una leyenda al final del informe de ensayo que indique "--Ultima Línea--"
15.5. Incumplimientos. Si se demuestra que el titular
comercializa el producto sin presentar al MS la Declaración de Conformidad
señalada en el numeral 15.4.1 del presente reglamento, el MS podrá solicitar el
retiro del mercado del producto en cuestión y la suspensión del registro
sanitario. En caso de que el titular del producto no presente la Declaración de
Conformidad en el plazo que 15 días naturales posterior a la notificación de
parte del MS del incumplimiento detectado, se procederá a la
cancelación del registro sanitario correspondiente. Además, si se detecta que
dos o más lotes de un producto medicinal a base de cannabis no cumplen con las
especificaciones establecidas en este reglamento técnico, ya sea en resultados
obtenidos como parte del análisis que se realice para la Declaración de
Conformidad o por controles en el mercado, el MS procederá con la cancelación
del registro sanitario correspondiente.
15.6. Reportes por parte de los fabricantes e importadores. Los
fabricantes e importadores deberán realizar un reporte anual durante el mes de
enero de cada año, con el listado de productos medicinales a base de cannabis
comercializados, el cual debe incluir la siguiente información por cada
producto: nombre del producto, número de registro sanitario, lote (s)
comercializado (s), fabricante o importador, número de informe de análisis
correspondiente a cada lote comercializado, pruebas llevadas a cabo ( en caso
de obtenerse un resultado no conforme deberá indicarse en el reporte señalado)
y nombre del laboratorio que realizó los análisis.
Dicha
información será suministrada al MS utilizando el correo electrónico dispuesto
por la institución en su página web o por medio del sistema de trazabilidad que
se establezca, donde se brindará un acuse de recibido consignando la fecha de
entrega y podrá ser confrontada en las visitas de seguimiento.
15.7. Verificación del cumplimiento del reglamento técnico. El
Ministerio de Salud debe verificar de manera aleatoria y representativa en los
puntos de venta o en las bodegas del fabricante nacional, importador o
comercializador, la conformidad de los productos regulados en este reglamento
técnico.
El
Ministerio de Salud para la verificación anterior, podrá realizar por su
cuenta, en convenio con otra organización o contratar Organismos de Evaluación
de Conformidad públicos o privados debidamente acreditados por el ECA o con
acreditación reconocida por el ECA, para que realicen inspecciones, ensayos o
verificaciones en el mercado.
Los
Organismos de Evaluación de Conformidad indicados en el párrafo anterior podrán
verificar en el mercado el cumplimiento de lo dispuesto en el presente
reglamento, para ello pueden:
15.
7 .1. Solicitar la documentación que sustenta el cumplimiento de
especificaciones de los lotes comercializados.
15.7.2.
Tomar muestras para efectuar ensayos relativos a la evaluación de la
conformidad.
16.- PUBLICIDAD Y PROHIBICIONES. Se
establecen las siguientes disposiciones relacionadas con la publicidad de
productos medicinales a base de cannabis.
16.1.
La publicidad de productos medicinales a base de cannabis debe ser aprobada
previamente por la DRPIS del MS y ser dirigida únicamente a los profesionales
de la salud, establecidos en el artículo 40 de la Ley Nº 5395 del 30 de octubre
de 1973 "Ley General de Salud".
16.1.2.
La publicidad de los productos medicinales a base de cannabis deberá incluir en
forma visual o auditiva, las siguientes leyendas:
16.1.2.1.
Este producto requiere receta médica.
16.1.2.2.
Indicar número de oficio de aprobación de la publicidad.
16.1.3.
El trámite, requisitos y plazo de resolución de las autorizaciones de la
publicidad será el establecido en los artículos 25 al 29 del Decreto Ejecutivo
Nº 36868-S del 12 de setiembre del 2011 "Reglamento para la autorización y
control sanitario de la publicidad de productos de interés sanitario".
16.2.
Se prohíbe auspicio o patrocinio y asignar bonificaciones de productos
medicinales a base cannabis a los pacientes, farmacias y profesionales en
ciencias de la salud.
16.3.
Queda prohibida la fabricación, importación, distribución y suministro en el
territorio nacional, de muestras sin valor comercial de productos medicinales a
base de cannabis.
17 .- TARIFAS. Se establecen las siguientes tarifas para los
trámites establecidos en el presente reglamento:

18.- AUTORIDADES COMPETENTES. Se establecen las
siguientes disposiciones respecto a las autoridades competentes.
18.1. Inspecciones y controles. La
verificación de este reglamento técnico y demás normativa aplicable lo
realizará el MS mediante inspecciones a los establecimientos, a fin de
garantizar la calidad y seguridad de estos productos. Asimismo, mediante los
controles en el mercado definidos en el numeral 15.7 del presente decreto.
18.2. Aplicación de medidas especiales. Si se
demuestra el incumplimiento de las disposiciones establecidas en el reglamento,
el MS aplicará las medidas especiales establecidas en el artículo 356 de la Ley
General de Salud, sin menoscabo de la responsabilidad civil o penal en que
hayan incurrido las personas fisicas o jurídicas responsables de tal
incumplimiento; y sin perjuicio de cualquier otra sanción conforme a la
legislación vigente.
19.- CONCORDANCIA. Este documento no coincide con
alguna norma internacional.
20.- BIBLIOGRAFÍA. Para la elaboración del presente
reglamento se utilizó como referencias bibliográficas las siguientes normas,
guías y directrices internacionales:
20.1. Autoridad Nacional de Medicamentos y Productos Sanitarios, IP
(INF ARMED) Portugal) (2019). Deliberación Nº 11CD2019.
20.2. INTECO (Instituto de Normas Técnicas de Costa Rica) (2017). Requisitos
generales para la competencia de laboratorios de ensayo y calibración (Norma
INTE/ISO/IEC 17025:2017).
20.3. INTECO (Instituto de Normas Técnicas de Costa Rica) (2020). Evaluación
de la conformidad- Vocabulario y principios generales (Norma INTE-ISO/IEC
17000:2020).
20.4. Farmacopea de los Estados Unidos Americanos y Formulario
Nacional. (2023). Capitulo <561> Artículos de origen botánico vigente
al 6 de octubre del 2023. Rockville, MD.
20.5. Farmacopea de los Estados Unidos Americanos y Formulario Nacional.
(2023). Capitulo <467> Disolventes residuales vigente al 6 de
octubre del 2023. Rockville, MD.
20.6. Ministry of Health New Zealand (2019). Misuse of Drugs
(Medicinal Cannabis) Regulations 2019 (LI 2019/321 ). disponible en:
https://www.legislation.govt.nz/regulation/public/2019/0321 /latest/whole.htm 1
20.7. Therapeutic Goods Administration (TGA). (2020). Microbiological
quality of medicinal cannabis products: Complying with Therapeutic Goods
(Microbiological Standards for Medicines) (TGO 100) Order 2018.
ANEXO I
(Normativo)
Formulario
de solicitud de notificación de material vegetal y derivados de cannabis
1.
Datos del producto.
1.1.
Nombre del producto.
1.2.
Nombre y concentración de los cannabinoides que contiene (CBD y/o THC).
1.3.
Forma farmacéutica.
1.4.
Presentación del producto.
1.5.
Vida útil propuesta.
1.6.
Descripción detallada del material del empaque primario
2.
Datos del fabricante (s) y acondicionador
2.1.
Nombre y país del o de los laboratorios que participen en la fabricación.
2.2.
Dirección, teléfono y correo electrónico.
2.3.
Etapa de fabricación en la que participa cada laboratorio.
2.4.
Número de permiso sanitario de funcionamiento y fecha de vencimiento (en caso
de ser nacional).
2.5
En caso de fabricantes nacionales número de licencia u autorización para la
fabricación de derivados de cannabis ( cáñamo o cannabis psi coactivo).
3.
Datos del titular del producto.
3.1.
Nombre.
3
.2. Dirección, teléfono y correo electrónico.
3.3.
País.
4.
Datos del representante legal.
4.1.
Nombre.
4.2.
Número de documento de identidad.
4.3.
Dirección, teléfono y correo electrónico.
5.
Datos de profesional responsable
5.1.
Nombre.
5.2.
Número de documento de identidad.
5.3.
Dirección, teléfono y correo electrónico.
5.4.
Número de colegiado.
ANEXOII
(Normativo)
Lista de
indicaciones terapénticas recomendadas para la prescripción de prodnctos
medicinales
a base de cannabis
a)
Dolor y espasticidad asociada a esclerosis múltiple o lesiones de la médula
espinal;
b)
Náuseas, vómitos (derivados de quimioterapia, radioterapia y combinación de
VIH, medicación para la hepatitis C);
c)
Estimulación del apetito en cuidados paliativos de pacientes sometidos a
tratamientos oncológicos o con SIDA;
d)
Dolor crónico (asociado a enfermedades oncológicas o del sistema nervioso, como
el dolor neuropatía causada por daño a los nervios, dolor del miembro fantasma,
neuralgia del trigémino o después del herpes zóster);
e)
Síndrome de Gilles de la Tourette;
f)
Epilepsia y tratamiento de trastornos convulsivos graves en la infancia, como
Dravet y Lennox-Gastaut;
g)
Glaucoma resistente a la terapia.
Nota: El
médico tratante de acuerdo con criterio profesional podrá prescribir productos
medicinales a base de cannabis en otras indicaciones siempre y cuando se
cumplan las siguientes condiciones:
. Si es
una enfermedad grave y se cumple alguno de los siguientes criterios:
a)
no existe una terapia estándar generalmente aceptada, o en casos particulares
la terapia estándar no se puede aplicar de acuerdo con la evaluación
justificada del médico tratante, considerando los efectos secundarios esperados
y el estado de la enfermedad del paciente, y/o;
b)
existe una posibilidad razonable de que el cannabis medicinal tenga un efecto
positivo en el proceso de la enfermedad o en síntomas graves, y/o;
c)
las terapias estándar han fallado.
ANEXO IIl
(Normativo)
Solicitud
para el registro sanitario (primera vez, renovación, cambios post registro),
Información
requerida.
1.
Datos del producto.
1.1
Nombre del producto.
1.2
Nombre y concentración de los cannabinoides que contiene (CBD y/o THC).
1.3
Forma farmacéutica.
1.4
Vía de administración.
1.5
Presentación del producto.
1.6
Vida útil propuesta.
1.
7. Descripción detallada del material del empaque primario
1.8
Tipos de solicitud (registro por primera vez, renovación, cambio post registro)
2.
Datos del fabricante (s) y acondicionador
2.1.
Nombre y país del o de los laboratorios que participen en la fabricación.
2.2.
Dirección, teléfono y correo electrónico.
2.3.
Etapa de fabricación en la que participa cada laboratorio.
2.4.
Número de permiso sanitario de funcionamiento y fecha de vencimiento, (cuando
sea nacional).
2.5.
En el caso de fabricante nacionales indicar número de licencia para la
fabricación de productos naturales medicinales a base de cannabis.
3.
Datos del titular del producto.
3.1.
Nombre.
3.2.
Dirección, teléfono y correo electrónico.
3.3. País.
4. Datos
del o los distribuidores.
4.1.
Nombre del o de los distribuidores.
4.2.
Dirección, teléfono y correo electrónico.
4.3.
Número de permiso sanitario y fecha de vencimiento.
5.
Datos del representante legal.
5.1.
Nombre.
5.2.
Número de documento de identidad.
5.3.
Dirección, teléfono y correo electrónico.
6.
Datos de profesional responsable
6.1.
Nombre.
6.2.
Número de documento de identidad.
6.3.
Dirección, teléfono, fax y correo electrónico.
6.4.
Número de colegiado.
7.
Leyenda que le dé carácter de declaración jurada en la solicitud.
4.
Datos del o los distribuidores.
ANEXO IV
(Normativo)
Requisitos
para la modificación al registro sanitario.
a)
Modificaciones que requieren aprobación previa del MS.
|
MODIFICACIÓN |
REQUISITOS |
|
1.
En la presentación comercial |
1.
Comprobante de pago. 2. Solicitud
firmada y sellada por el profesional responsable. 3.
Nuevas etiquetas originales del envase/empaque primario, secundario o sus
proyectos según de acuerdo con lo establecido en el numeral 12.2 y 12.3 del
presente reglamento. 4.
Documentos emitidos por el titular o su representante legal que declare el
cambio. |
|
2.
En el nombre del producto |
l.
Comprobante de pago. 2.
Solicitud firmada y sellada por el profesional responsable. 3. Documentos
emitidos por el titular o su representante legal que declare el cambio de
nombre. 4.
Nuevas etiquetas originales del envase/empaque primario, secundario acuerdo
con lo establecido en los numerales 12.2 y 12.3 del presente reglamento. |
|
3.
Razón social del fabricante empacador o titular |
1.
Comprobante de pago. 2.
Solicitud firmada y sellada por el profesional responsable. 3.
Documento legal que acredite el cambio. 4.
Nuevas etiquetas originales del envase/empaque primario, secundario o sus
proyectos de acuerdo con lo establecido en el numeral 12.2 y 12.3 del
presente reglamento |
|
4.
En la ficha técnica |
1.
Comprobante de pago. 2.
Solicitud firmada y sellada por el profesional responsable. 3.
Documento legal que acredite el cambio. 4.
Nuevas etiquetas originales del envase/empaque primario, secundario o sus
proyectos de acuerdo con lo establecido en los numerales 12.2 y 12.3 del
presente reglamento. |
|
5.
En el período de vida útil |
l.
Comprobante de pago. 2.Solicitud
firmada y sellada por el profesional responsable. 3.Informe
de estudio de estabilidad que confirme el período de vida útil propuesto. Ver
nota 1 del numeral 10.2.13 del presente reglamento. 4.Documento
emitido por el titular o representante legal que acredite el cambio. |
|
6.
En las condiciones de almacenamiento. |
1.
Comprobante de pago. 2.Solicitud
firmada y sellada por el profesional responsable. 3.Informe
de estudio de estabilidad que respalde las condiciones solicitadas Ver nota 1
del numeral 10.2.13 del presente reglamento. 4.Nuevas
etiquetas originales del envase/empaque primario, secundario o sus proyectos
de acuerdo con lo establecido en los numerales 12.2 y 12.3 del presente
reglamento. 5.Documento
emitido por el titular o representante legal que acredite el cambio. |
|
7. De
empacador primario |
1.
Comprobante de pago. 2.
Solicitud firmada y sellada por el profesional responsable. 3.Certificado
de buenas prácticas de manufactura de nuevo empacador. 4.
Contrato con el nuevo empacador, en caso de fabricaciones por terceros. 5.Nuevas
etiquetas originales del envase/empaque primario, secundario o sus proyectos
de acuerdo con lo establecido en los numerales 12.2 y 12.3 del presente
reglamento. 6.
Declaración jurada en la que manifieste que se mantienen las mismas
condiciones referente a la composición o fórmula cualicuantitativa, tipo y
material de empaque primario, proceso y lugar de manufactura del producto
registrado. 7.
Documento emitido por el titular o representante legal que acredite el
cambio. |
|
8.
Tipo de material del empaque primario o del sistema envase-cierre. |
l.
Comprobante de pago. 2.
Solicitud firmada y sellada por el profesional responsable. 3.
Informe de estudio de estabilidad. Ver nota I del numeral 10.2.13 del
presente reglamento. 4. Especificaciones
del empaque primario o sistema de envase - cierre. 5.
Documento emitido por el titular o representante legal que acredite el
cambio. |
|
9.
Adición de un nuevo empaque primario |
l.
Comprobante de pago. 2.
Solicitud firmada y sellada por el profesional responsable. 3.
Informe de estudio de estabilidad para el empaque solicitado. 4.
Nuevas etiquetas originales del envase/empaque primario, secundario o sus
proyectos de acuerdo con lo establecido en el numeral 12.2 y 12.3 del
presente r:eglamento. 5.
Especificaciones del empaque primario. 6.
Documento emitido por el titular o representante legal que acredite el
cambio. |
|
10.
De titular |
l.
Comprobante de pago. 2.
Solicitud firmada y sellada por el profesional responsable. 3.
Nuevas etiquetas originales del envase/empaque primario, secundario o sus
proyectos de acuerdo con lo establecido en los numerales 12.2 y 12.3 del
presente reglamento. 4.
Documento legal que acredite el cambio adjuntando los nuevos poderes 5.
Contrato de conformidad con el numeral 10.2.7 de este reglamento en caso de
fabricación por terceros. |
|
11.
En caso de fabricación por terceros: a)
Cambio de fabricante. b)
Cambio de fabricante y de país de origen |
1.
Comprobante de pago. 2.Solicitud
firmada y sellada por el profesional responsable. 3.
Certificado de la venta del producto. 4.
Nuevas etiquetas originales del envase/empaque primario, secundario o sus
proyectos de acuerdo con lo establecido en los numerales 12.2 y 12.3 del
presente reglamento. 5.
Certificado de buenas prácticas de manufactura del nuevo fabricante. 6.
Informe de estudio de estabilidad. Ver nota 1 del numeral 10.2.13 del
presente reglamento. 7.
Metodología analítica. 8.
Contrato con el nuevo fabricante de conformidad al numeral I 0.2.7 del presente
reglamento. 9.
Documento emitido por el titular o representante legal que acredite el
cambio. |
|
12.
Cambio de excipientes o cambio en la concentración de estos. |
1.
Comprobante de pago. 2.
Solicitud firmada y sellada por el profesional responsable. 3.
Fórmula cuali-cuantitativa por unidad de dosis 4.
Informe de estudio de estabilidad. Ver nota 1 del numeral 10.2.13 del
presente reglamento. 5.
Justificación técnica del cambio. 6.
Metodología analítica del producto terminado actualizadas, cuando aplique. 7.
Documento emitido por el titular o representante legal que acredite el
cambio. |
|
13.
Información en el etiquetado primario y secundario |
1.
Comprobante de pago. 2.Solicitud
firmada y sellada por el profesional responsable. 3.Nuevas
etiquetas originales del envase/empaque primario, secundario o sus proyectos
de acuerdo con lo establecido en los numerales 12.2 y 12.3 del presente
reglamento. 4.Justificación
técnica del cambio emitida por el titular o su representante legal. |
|
14.
Sitio de fabricación dentro del mismo país |
1.
Comprobante de pago. 2.Solicitud
firmada y sellada por el profesional responsable. 3.Certificado
de buenas prácticas de manufactura. 4.
Declaración jurada del titular del producto o representante legal donde haga constar
que las condiciones de fabricación han variado. 5.Documento
emitido por el titular o su representante legal que acrediten el cambio. |
|
15.
De representante legal o del profesional responsable |
l.
Comprobante de pago. 2.Solicitud
firmada y sellada por el profesional responsable. 3.
Poder otorgado de acuerdo con la legislación de cada estado Parte que
acredite el cambio. |
b) Modificaciones que deben notificarse al MS y no
requieren aprobación previa.
|
MODIFICACIÓN |
REQUISITOS |
|
l.
Material o dimensiones del empaque secundario |
l.
Notificación firmada y sellada por el profesional responsable. 2.
Documento emitido por el titular o su representante legal que declare el
cambio. 3.
Empaque originales o sus proyectos de acuerdo con lo establecido en los
numerales 12.2 y 12.3 del presente reglamento. |
|
2.
Diseño del etiquetado del empaque primario |
l.
Notificación firmada y sellada por el profesional responsable. 2.
Documento emitido por el titular o su representante legal que declare el cambio.
3.
Empaque originales o sus proyectos. |
|
3.
Descontinuación de presentaciones registradas |
1.
Notificación firmada y sellada por el profesional responsable. 2.
Documento emitido por el titular o su representante legal que declare el
cambio. |
|
4. Cambio
o ampliación de distribuidor |
l.
Notificación firmada y sellada por el profesional responsable. 2.
Solicitud firmada y sellada por el profesional responsable. 3.
Documento legal emitido por el titular o su representante legal avale el
cambio o la ampliación. |
ANEXO V
(Normativo)
FORMATO DE DECLARACIÓN
DE CONFORMIDAD
DECLARACIÓN DE
CONFORMIDAD No. (Colocar identificación única y clara en
donde se le asigne un número
de serie o codificación unívoca)
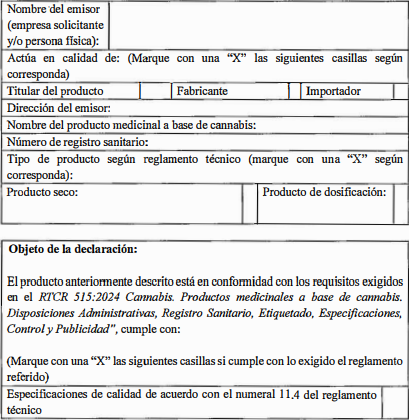
Información
adicional
Como
soporte de esta declaración de conformidad, se adjunta la siguiente
información:

Observaciones: Declaro bajo fe de juramento que la información aportada
para esta solicitud es fiel y verdadera. Asimismo, declaro conocer que de no
decir la verdad incurro en el delito de perjurio, sancionado por el Código
Penal. Finalmente, soy conocedor de que sí la autoridad llegase a corroborar
alguna falsedad en la presente declaración, errores u omisiones en los
documentos aportados deberé asumir las sanciones establecidas por la Ley y que,
en todó caso, los documentos adjuntos están a disposición de la Autoridad
Competente, en el domicilio indicado arriba.
